
A convenient method for preparation of pure standards of peroxyacetyl nitrate for atmospheric...
4
Atmospheric Enrironmem VoL 16, No 10. pp. 2447 2450. 1982 CO34 6981 82 102447 04 103.00/0 Printed in Great Britain ( 1982 Pergamon Press Lid • A CONVENIENT METHOD FOR PREPARATION OF PURE STANDARDS OF PEROXYACETYL NITRATE FOR ATMOSPHERIC ANALYSES TORBEN NIELSEN Chemistry Department, Rise National Laboratory, DK-4000 Roskilde, Denmark ANNE MARIA HANSEN and E~LING LUND THOMSEN Air Pollution Laboratory, National Agency of Environmental Protection, Rise National Laboratory, DK-4000 Roskilde, Denmark {First received I September 1981 and in finalJorm 27 November 1981) Abstraet--Peroxyacetyl nitrate (PAN) is synthesized by nitration of peracetic acid (1.2 M), extracted by n- heptane, and purified with normal-phase high-performance liquid chromatography. The purified PAN solution is free of acetyl nitrate. The content of PAN is determined by means of hydrolysis of PAN into nitrite, and determination by ion chromatography of nitrite and nitrate (formed by oxidation of nitrite). The purified PAN solution is used for the calibration of the gas chromatograph with electron capture detection. I. INTRODUCTION Peroxyacetyl nitrate (PAN) is formed in the atmos- phere by the action of solar radiation on a mixture of hydrocarbons and nitrogen oxides (NAS, 1977; Stephens, 1969)and may even be present in relatively high concentrations in photochemically polluted air after long-range transport (Nielsen et al., 1981). PAN seems to be the most useful indicator for the charac- terization of photochemical air pollution (NAS, 1977; Stephens 1969). Its determination is usually performed by means of gas chromatography using electron capture (EC) detection (Penkett et al., 1975; Stephens, 1969), but the absolute calibration of the detector presents severe problems• The most attractive approach is probably the coulometric method (Lovelock et al., 1971) which, once the coulometric response is established, requires no calibration .¢amples at all. Even if PAN, due to high electron capture rate constant and absence of electron capturing reaction products in the EC detector, lent itself to coulometric calibration, this method is un- suitable, as PAN partly decomposes during the gas chromatographic separation (Sandalls et al., 1974). It appears to be difficult and inconvenient to determine and control the extent of the decomposition processes• Beyond the general problems connected with the preparation of calibration samples for the EC detector, PAN represents specific problems due to its high reactivity and dangerous handling. Pure liquid PAN is extremely explosive, and storage of the PAN standard as a highly diluted gas is unsatisfactory, since conden- sation of PAN in storage at low temperatures increases the risk of explosions, and PAN is not stable for longer periods at higher temperatures 10-15' C) (Bruckmann and MOlder. 1979; Stephens, 1969). A convenient procedure for the preparation of PAN standards via the photochemical chlorine-aldehyde reaction (Gay et al., 1976)is only of practical value for the limited number of laboratories having highly sensitive infrared spectroscopy equipment. Two pro- cedures for the condensed phase synthesis of PAN have been described, both based upon the nitration of 40 ','.,, peracetic acid (Hendry and Kenley, 1977; Kravetz et al., 1980). But this is not commercially available in some countries; neither is 9030 hydrogen peroxide, which may be used for the preparation of 40°.,, per- acetic acid (Greenspan, 1946). Furthermore, the con- centration of 30~o hydrogen peroxide to 9030 (Hurd and Puterbaugh, 1930) caused a serious explosion in our laboratories. The procedure reported here for the preparation of PAN standards for calibration of the EC detector describes the synthesis of PAN in solution by means of standard chemicals, the purification of the PAN solutions by means of high-performance liquid chro- matography (HPLC), the determination of the PAN content in the purified solutions by means of ion- chromatography (IC) and the characterization of the synthesized PAN. The advantages of the method are: low risks, if any, of explosions; the PAN solutions can be stored for months without any observable decomposition and the high purity of the PAN solutions excludes the possibility of interfering impurities. Furthermore, the procedures described here do not require any specific knowledge or equipment, which is not available in most laboratories investigating air pollution. 2447
-
Upload
torben-nielsen -
Category
Documents
-
view
216 -
download
4
Transcript of A convenient method for preparation of pure standards of peroxyacetyl nitrate for atmospheric...

Atmospheric Enrironmem VoL 16, N o 10. pp. 2447 2450. 1982 CO34 6981 82 102447 04 103.00/0 Printed in Great Britain ( 1982 Pergamon Press Lid
• A C O N V E N I E N T M E T H O D FOR PREPARATION OF PURE S T A N D A R D S OF PEROXYACETYL NITRATE FOR
ATMOSPHERIC ANALYSES
TORBEN NIELSEN Chemistry Department, Rise National Laboratory, DK-4000 Roskilde, Denmark
ANNE MARIA HANSEN and E~LING LUND THOMSEN Air Pollution Laboratory, National Agency of Environmental Protection, Rise National Laboratory,
DK-4000 Roskilde, Denmark
{First received I September 1981 and in f ina lJorm 27 November 1981)
Abstraet--Peroxyacetyl nitrate (PAN) is synthesized by nitration of peracetic acid (1.2 M), extracted by n- heptane, and purified with normal-phase high-performance liquid chromatography. The purified PAN solution is free of acetyl nitrate. The content of PAN is determined by means of hydrolysis of PAN into nitrite, and determination by ion chromatography of nitrite and nitrate (formed by oxidation of nitrite). The purified PAN solution is used for the calibration of the gas chromatograph with electron capture detection.
I. INTRODUCTION
Peroxyacetyl nitrate (PAN) is formed in the atmos- phere by the action of solar radiation on a mixture of hydrocarbons and nitrogen oxides (NAS, 1977; Stephens, 1969)and may even be present in relatively high concentrations in photochemically polluted air after long-range transport (Nielsen et al., 1981). PAN seems to be the most useful indicator for the charac- terization of photochemical air pollution (NAS, 1977; Stephens 1969). Its determination is usually performed by means of gas chromatography using electron capture (EC) detection (Penkett et al., 1975; Stephens, 1969), but the absolute calibration of the detector presents severe problems•
The most attractive approach is probably the coulometric method (Lovelock et al., 1971) which, once the coulometric response is established, requires no calibration .¢amples at all. Even if PAN, due to high electron capture rate constant and absence of electron capturing reaction products in the EC detector, lent itself to coulometric calibration, this method is un- suitable, as PAN partly decomposes during the gas chromatographic separation (Sandalls et al., 1974). It appears to be difficult and inconvenient to determine and control the extent of the decomposition processes•
Beyond the general problems connected with the preparation of calibration samples for the EC detector, PAN represents specific problems due to its high reactivity and dangerous handling. Pure liquid PAN is extremely explosive, and storage of the PAN standard as a highly diluted gas is unsatisfactory, since conden- sation of PAN in storage at low temperatures increases the risk of explosions, and PAN is not stable for longer
periods at higher temperatures 10-15' C) (Bruckmann and MOlder. 1979; Stephens, 1969). A convenient procedure for the preparation of PAN standards via the photochemical chlorine-aldehyde reaction (Gay et al., 1976)is only of practical value for the limited number of laboratories having highly sensitive infrared spectroscopy equipment. Two pro- cedures for the condensed phase synthesis of PAN have been described, both based upon the nitration of 40 ','.,, peracetic acid (Hendry and Kenley, 1977; Kravetz et al., 1980). But this is not commercially available in some countries; neither is 9030 hydrogen peroxide, which may be used for the preparation of 40°.,, per- acetic acid (Greenspan, 1946). Furthermore, the con- centration of 30~o hydrogen peroxide to 9030 (Hurd and Puterbaugh, 1930) caused a serious explosion in our laboratories.
The procedure reported here for the preparation of PAN standards for calibration of the EC detector describes the synthesis of PAN in solution by means of standard chemicals, the purification of the PAN solutions by means of high-performance liquid chro- matography (HPLC), the determination of the PAN content in the purified solutions by means of ion- chromatography (IC) and the characterization of the synthesized PAN.
The advantages of the method are: low risks, if any, of explosions; the PAN solutions can be stored for months without any observable decomposition and the high purity of the PAN solutions excludes the possibility of interfering impurities. Furthermore, the procedures described here do not require any specific knowledge or equipment, which is not available in most laboratories investigating air pollution.
2447
24.48 TORBEN NIELSON el al.
2. SYNTHESIS
We considered that the nitration of peracetic acid in solution is only possible if the content of water is low. By making the peracetic acid from 30yo hydrogen peroxide, we used an appropriate amount of acetic acid anhydride instead of acetic acid (Greenspan, 1946) in other to remove the water: 55 ml acetic acid anhydride (0.58 mol, freshly distilled) was cooled to c. 10°C. 0.5 ml sulphuric acid (suprapur, Merck) was added. 10ml c. 30 Oo hydrogen peroxide (p.a. Merck, c. 0.1 mol hydro- gen peroxide and c. 0.4 mol water) was added at this temperature with stirring within 1 h. The solution was kept at room temperature until next day. After 21 h, the concentration of hydrogen peroxide was 0.006 M and the concentration of peracetic acid was 1.2 M (Swern, 1953). The peracetic acid solution was stored at 4°C. Precautions such as the use of clean glass equipment and avoiding contact between the peroxide solutions and metal surfaces should, of course, be taken.
The PAN was synthesized as follows: A 250ml 3- necked flask equipped with a thermometer and a magnetic stirring bar was charged with 100ml n- heptane (Uvasol Merck). The flask was placed in a 2- propanol bath, and its content was stirred while sufficient dry ice was added to the bath to cool the n- heptane to 0°C. 10.0 ml peracetic acid solution [1.2 M) was added with stirring at this temperature, followed by concentrated sulphuric acid (8.0ml, Merck supra- pur) and within 5min 2.0ml nitric acid (65~0, p.a. Merck) was slowly added. After stirring at 0°C for 5min, the mixture was poured on 100g ice water in a separatory funnel. The n-heptane phase containing the PAN was separated from the water phase and stored at - 15°C. Standing at these conditions, the loss of PAN, if any, during a period of 3 months was low. The yields of PAN were 15-24~,~i (2-4mgml-~}.
Repeating the washing of the organic phase with ice water (Kravetz et al., 1980) led to lower yield of PAN. Performing the synthesis at more acidic conditions may perhaps give a higher yield of PAN. But since the described procedure gives much more PAN than required for the calibrations during one summer, no attempts were made to optimize the yield.
3. PURIFICATION BY H P L C
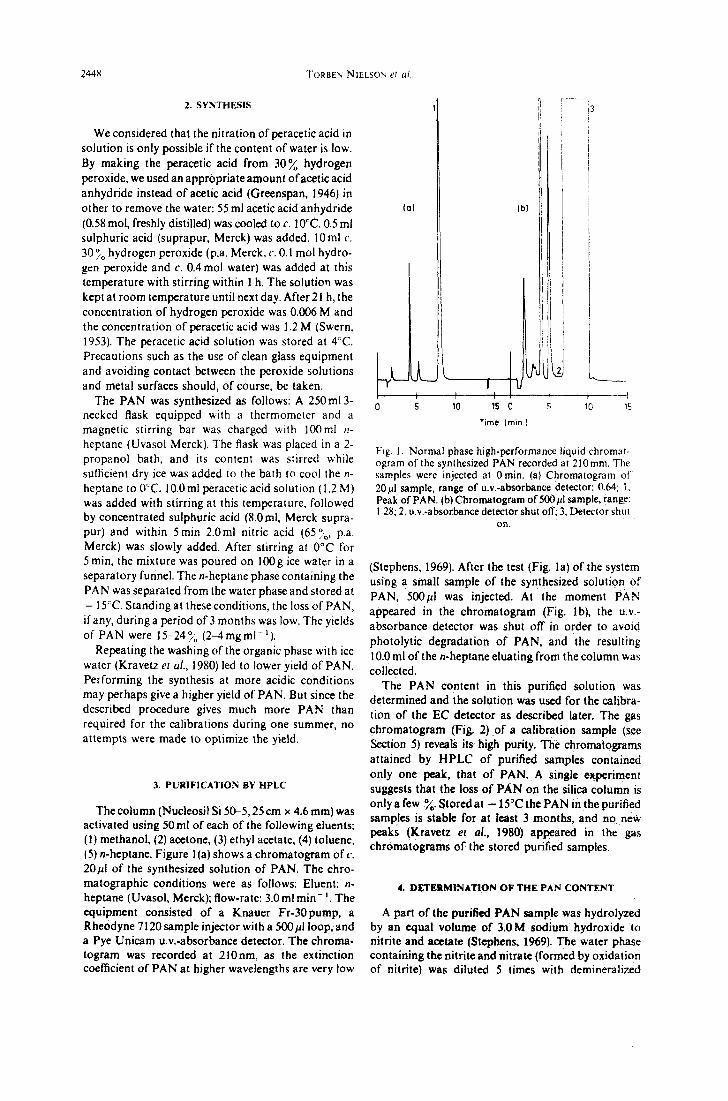
The column (Nucleosil Si 50-5, 25 c m x 4.6 ram) was activated using 50ml of each o f the following eluents: (1) methanol, (2) acetone, (3) ethyl acetate, (4) toluene, (5) n-heptane. Figure 1 (a) shows a chromatogram of c. 20/21 of the synthesized solution of PAN. The chro- matographic conditions were as follows: Eluent: n- heptane (Uvasol, Merck); flow-rate: 3.0ml min- '. The equipment consisted of a Knauer Fr-30pump, a Rheodyne 7120 sample injector with a 500/21 loop, and a Pye Unicam u.v.-absorbance detector. The chroma- togram was recorded at 210rim, as the extinction coefficient of P A N at higher wavelengths are very low
{a)
o 5
i . . . . . . . .
! 13
I I I t ' I 10 15 0 !S 10 15
T i m e ( r a i n . }
Fig. 1. Normal phase high-performance liquid chromat- ogram of the synthesized PAN recorded at 210 ram. The samples were injected at 0 rain. (a} Chromatogram of 20/21 sample, range of u.v.-absorbance detector: 0.64; 1. Peak of PAN. {b) Chromatogram of 500/21 sample, range: 1.28; 2, u.v.-absorbance detector shut off; 3, Detector shut
on.
(Stephens, 1969). After the test (Fig. la) of the system using a small sample of the synthesized solution of PAN, 500/21 was injected. At the moment PAN appeared in the chromatogram (Fig. lb), the u.v.- absorbance detector was shut off in order to avoid photolytic degradation of PAN, and the resulting 10.0 ml of the n-heptane eluating from the column was collected.
The PAN content in this purified solution was determined and the solution was used for the calibra- tion of the EC detector as described later. The gas chromatogram (Fig. 2} of a calibration sample (see Section 5) reveals its high purity. The chromatograms attained by HPLC of purified samples contained only one peak, that of PAN. A single experiment suggests that the loss of PAN on the silica column is only a few %. Stored at - 15°C the PAN in the purified samples is stable for at least 3 months, and no new peaks {Kravetz et al., 1980) appeared in the gas chromatograms of the stored purified samples.
4. D E T E R M I N A T I O N O F T H E PAN C O N T E N T
A part of the purified PAN sample was hydrolyzed by an equal volume of 3.0 M sodium hydroxide to nitrite and acetate (Stephens, 1969}. The water phase containing the nitrite and nitrate (formed by oxidation of nitrite) was diluted 5 times with demineralized
A convenient method for preparation of pure standards of peroxyacetyl nitrate for atmospheric analyses 2449
3
I I I I I I , . ,
28 2/. 20 16 12 8 /. 0
Time (rain.)
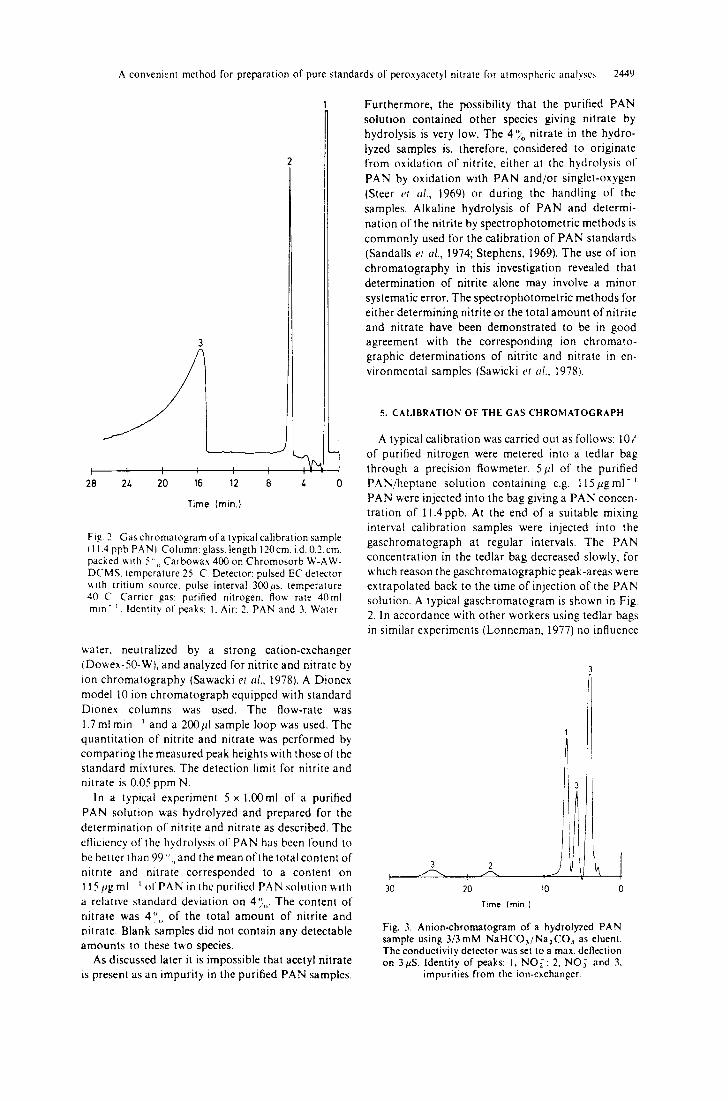
Fig. 2. Gas chromatogram of a typical calibration sample l11.4 ppb PAN L Column: glass, length 120 cm, i.d. 0.2. cm. packed v, ith 5 ",, Carbowax 400 on Chromosorb W-AW- DCMS. temperature 25 C. Detector: pulsed EC detector v, ith tritium source, pulse interval 300ps. temperature 40 C. Carrier gas: purified nitrogen, flow rate 40ml rain- ~. Identity of peaks: 1, Air: 2. PAN and 3, Water.
water, neutralized by a strong cation-exchanger (Dowex-50-WL and analyzed for nitrite and nitrate by ion chromatography (Sawacki et al., 1978). A Dionex model 10 ion chromatograph equipped with standard Dionex columns was used. The flow-rate was 1.7 ml min- J and a 200,ul sample loop was used. The quanti tat ion of nitrite and nitrate was performed by comparing the measured peak heights with those ot the standard mixtures. The detection limit for nitrite and nitrate is 0.05 ppm N.
In a typical experiment 5 x 1.00ml of a purified PAN solution was hydrolyzed and prepared for the determination of nitrite and nitrate as described. The efficiency of the hydrolysis of PAN has been Iound to be better than 99 ",, and the mean of the total content of nitrite and nitrate corresponded to a content on 115pgml ~ of PAN m lhe purilicd PAN solution with a relative standard deviation on 4'),,. The content of nitrate was 4'~,, of the total amount of nitrite and nitrate. Blank samples did not contain any detectable amounts to these two species.
As discussed later it is impossible that acetyl nitrate is present as an impurity in the purified PAN samples.
Furthermore, the possibility that the purified PAN solution contained other species giving nitrate by hydrolysis is very low. The 490 nitrate in the hydro- lyzed samples is, therefore, considered to originate from oxidation of nitrite, either at the hydrolysis of PAN by oxidation with PAN and/or singlet-oxygen (Steer e t a / . , 1969) or during the handling of the samples. Alkaline hydrolysis of PAN and determi- nation of the nitrite by spectrophotometric methods is commonly used for the calibration of PAN standards (Sandalls et al., 1974; Stephens, 1969). The use of ion chromatography in this investigation revealed that determination of nitrite alone may involve a minor systematic error. The spectrophotometric methods for either determining nitrite or the total amount of nitrite and nitrate have been demonstrated to be in good agreement with the corresponding ion chromato- graphic determinations of nitrite and nitrate in en- vironmental samples (Sawicki et al., 19781.
5. CALIBRATION OF THE GAS CHROMATOGRAPH
A typical calibration was carried out as follows: 10 g of purified nitrogen were metered into a tedlar bag through a precision ftowmeter. 5pl of the purified PAN/heptane solution containing e.g. l l 5 p g m 1 - 1 PAN were injected into the bag giving a PAN concen- tration of 11.4 ppb. At the end of a suitable mixing interval calibration samples were injected into the gaschromatograph at regular intervals. The PAN concentration in the tedlar bag decreased slowly, for which reason the gaschromatographic peak-areas were extrapolated back to the time of injection of the PAN solution. A typical gaschromatogram is shown in Fig. 2. In accordance with other workers using tedlar bags in similar experiments (Lonneman, 1977) no influence
3 2 , /
30 20 10 0
Time (rain,)
Fig. 3. Anion-chromatogram of a hydrolyzed PAN sample using 3/3mM NaHCOa/Na2CO ~ as eluent. The conductivily detector was set to a max. deflection on 3MS. Identity of peaks: 1, NO.~: 2, NO~ and 3,
impurities from the ion-exchanger.

2450 TORBEN NIELSEN et al.
of the dilution gas humidity was noticed. The es- timated calibration accuracy including the ionchro- matographic uncertainty, is 9 9'0-
6. CHARACTERIZATION
assume that acetyl nitrate is destructed on the silica column.
Acknowledgements--The skilful technical assistance of Bente Christensen, Helge Egsgaard and Ole John Nielsen, Chemistry Department, is grateful acknowledged.
No single test can give an appropriate evidence for the identity of PAN, if this is present in a diluted solution (Stephens et al., 1961, Stephens, 1969). Several experiments were performed in order to characterize the synthesized product. These confirmed, that the synthesized compound was PAN, and the high purity of the purified product. The results are described in the following:
(1) The infrared spectra of the synthesized solution and the purified solution showed a strong band at 1735cm - I and a band at 1836em -1 (uncorrected values) in agreement with the vapour phase spectrum of PAN (Stephens, 1969). These bands were not present in the blank samples.
(2) The field ionization mass spectrum of the syn- thesized solution showed the two peaks at m/z = 43 and 46, with that at m/z = 43 as the most abundant. No peaks corresponding to the molecular ion, m/z = 122 was observed. This spectrum in accordance with that of PAN reported elsewhere (Stephens, 1969).
(3) The chrornatograms of the synthesized PAN attained by HPLC at different wavelengths showed that PAN had one band with a max. below 210 rim, and no bands at higher wavelengths. This agrees with the vapour phase u.v.-spectrum of PAN (Stephens, 1969).
(4) The synthesized product and the purified PAN was able to oxidize iodide to iodine. None of the other components in the chromatogram (Fig. la) showed oxidizing properties.
(5) The purified product was unstable at room temperature and reacts instantaneous with sodium hydroxide solutions giving nitrite, as expected for PAN (Stephens, 1969).
(6) The retention time of the purified product had exactly the same retention time as that of PAN formed by irradiation of a mixture of trans-2-butene and nitrogen dioxide (Fild and Lovell, 1970) by GC with EC detection on two different columns, respectively, 5 ~ Carbowax 400 on Chromosorb W - A W - D C M S (Sandalls et al., 1974; Stephens, 1969) (Fig. 2) and 5 ~o Q F - I and 0.1 ~o diglycerol on Chromosorb W - A W - D C M S (Nieboer and van Ham, 1976).
(7) The fact, that the purified product gave only one peak at, respectively, normal phase HPLC and GC on two different columns, shows, that the possibility of the presence of any interfering impurities in this product is very low.
(8) None of the experiments described so far ex- cludes totally that the purified PAN contains c. 4 9o acetyl nitrate compared to PAN. Acetyl nitrate was, therefore, synthesized (Sifniades, 1975) and extracted with n-heptane. This solution containing 2000ppm acetyl nitrate gave no acetyl nitrate peak on HPLC using the same conditions as described for PAN. We
REFERENCES
Bruckmann P. and MOlder W. (1979) Die Messung yon Peroxiacetylnitrat (PAN) in der Aussenluft--Verfahren und erste Ergebnisse. Schriftenreihe der Landesanstalt fur Immissionssehutz der I.andes Nordrhein-West falen, Essen 47, 30-41.
Fild I. and Lovell D. J. (1970) Infrared spectral studies of peroxyacetyl nitrate (PAN). J. opt. Soc. A m. 60, 1315 ~ 1318.
Gay, B. W., Jr. Noonan R. C., Bufalini J. J. and Hanst P. L. (1976) Photochemical synthesis of peroxyacyl nitrates in gas phase via chlorine-aldehyde reaction. Envir. Sci Technol. !0, 82-85.
Greenspan F. P. (1946} The convenient preparation of per- acids. J. Am Chem. Soc. 6g, 907.
Hendry C. D. and Kenley R. A. (1977) Generation of peroxy radicals from peroxy nitrates (RO2NO2). Decomposition of pcroxyacyl nitrates. J. Am. Chem. Sac. 99, 3198-3199.
Hurd C. D.and l%terhaugh M. P. (1930) Concentration of hydrogen peroxide solutions. J. Am. Chem. Soc. 52, 950-953.
Kravetz T. M., Martin S. W. and Mendenhall G. D. (1980) Synthesis of perosyaeetyl and peroxyaroyl nitrates. Complexation of pcroxyacetyl nitrate with benzene. Envir. Sci. Technol. 14, 1262-1264.
Lonneman W. A. (1977) PAN measurement in dry and humid atmospheres. Envir. Sci. Technol. 11, 194-195.
Lovelock J. E., Maggs R. 5. and Adlard E. R. (1971) Gas-phase coul0metry by thermal electron attachment. Analyt. Chem. 43, 1962-1965.
National Academy of Sciences (1977). Ozone and other photochemical oxidants, NAS, Washington D.C.
Niebaer H. and van Ham J. (1976) Peroxyacetyl nitrate (PAN) in relation to ozone and some meteorolo~l parameters at Delft in the Netherlandsl Atmospheric Environment 10, 115-120.
Nielsen T., Samuelsson U., Grennfelt P, and Thomsen E. L. (1981) Peroxyacetyl nitrate in long-range transported polluted air. Nature 293, 553-555.
Penkett S. A., Sandalls F. J. and Lovelock J. E. (1975) Observations of peroxyacetyl nitrate (PAN) in air in southern England. Atmospheric Environment 9, 139-140.
Sandalls F. J., Penkett S. A. and Jones B. M. R. (1974) Preparation of peroxya~tyl nitrate (PAN) and its determi, nation in the atmosphere. AERE Report R 7807, HMSO, London.
Sawicki E., Mulik J. D. and Wittgenstein E. (1978) Ion Chromotooraphic Analysis of Environmental Pollutants. Ann Arbor, Ann Arbor, MI.
Sifniades S. (1975) Nitration of acetoaeetate esters by acetyl nitrate. A high yield synthesis of nitroacetoaeetate and nitroacetate esters. J. org. Chem. 40, 3562-3566.
Steer R. P., Darnall K. R. and Pitts J. N., Jr. (i969) The base- induced decomposition of peroxyaeetyl nitrate. Tetrahedron Lett. 11, 3765-3767.
Stephens E, R., Darley E. F., Taylor E. C. and Scott W. E (1961) Photochemical reaction products in air pollution. Int. J. Air War. Pollut. 4, 79-100.
Stephens E. R. (1969) The formation, reactions and pro- perties of peroxyacyi nitrates (PANs) in photochemical air pollution. Adv. Envir. Sci. Technol. 1, 119--146,
Swern D. (19.53) Epoxidation and hydroxylation of ethylenic compounds with organic peracids. In Oroanic Reactions VII (Edited by Adams R., Blatt A. H., Cope A. C., McGrew F. C., Niemann C. and Snyder H. R., pp. 378--433. John Wiley, New York.


















