
A Continuous Optimization Approach to Protein Design …suresh/Theses/SouravRakshitThesis.pdf · A...
201
A Continuous Optimization Approach to Protein Design with Structural and Functional Constraints A Thesis Submitted for the Degree of Doctor of Philosophy in the Faculty of Engineering By Sourav Rakshit DEPARTMENT OF MECHANICAL ENGINEERING INDIAN INSTITUTE OF SCIENCE BANGALORE - 560 012 INDIA April 2011
Transcript of A Continuous Optimization Approach to Protein Design …suresh/Theses/SouravRakshitThesis.pdf · A...

A Continuous Optimization Approach toProtein Design with Structural and
Functional Constraints
A Thesis
Submitted for the Degree of
Doctor of Philosophy
in the Faculty of Engineering
By
Sourav Rakshit
DEPARTMENT OF MECHANICAL ENGINEERING
INDIAN INSTITUTE OF SCIENCE
BANGALORE - 560 012
INDIA
April 2011

Contents
ii

i
To
The loving memory of
My grandparents
Binapani Rakshit
Sudhir Chandra Rakshit
Padmabati Saha
Sudhir Kumar Saha

ii
Table of Contents Title Page number
Abstract ................................................................................................................. iv
Acknowledgments ................................................................................................. v
List of Figures ...................................................................................................... vii
List of Tables ........................................................................................................ xi
List of Equations .................................................................................................. xii
1. Introduction ........................................................................................................ 1
1.1 Preamble ...................................................................................................... 1
1.2 Proteins ........................................................................................................ 3
1.2.1 A brief overview of protein structure and folding ................................ 3
1.2.2 A brief overview of protein design ...................................................... 8
1.3 Motivation .................................................................................................. 12
1.4 Problem statement ..................................................................................... 14
1.5 Scope of the thesis ..................................................................................... 17
1.6 Organization of the thesis .......................................................................... 18
1.7 Closure ....................................................................................................... 19
2. Literature Review ............................................................................................. 20
2.1 Reduced amino acid alphabet ................................................................... 20
2.2 Computational protein sequence design ................................................... 22
2.3 Elastic networks ....................................................................................... 25
2.4 Minimalist coarse-grained models ........................................................... 28
2.5 Closure ...................................................................................................... 31
3. Reduced Amino Acid Alphabet using Metric Multi-dimensional Scaling ...... 32
3.1 Introduction ............................................................................................... 32
3.2 Method ...................................................................................................... 33
3.3 Results and discussion ............................................................................... 37
3.4 Closure ....................................................................................................... 44
4. Search in the Sequence Space .......................................................................... 45
4.1 Introduction ............................................................................................... 45
4.2 The Double Sigmoid method ..................................................................... 49

iii
4.2.1 Formulation of the continuous optimization problem ........................ 49
4.2.2 Formulation of the constraints ............................................................ 55
4.2.3 Results ................................................................................................ 55
4.3 The Quadratic Programming method ........................................................ 69
4.3.1 Method ............................................................................................... 69
4.3.2 Results ................................................................................................ 71
4.4 Discussion .................................................................................................. 79
4.5 Closure ....................................................................................................... 81
5. Search in the Conformation Space ................................................................... 82
5.1 Introduction ............................................................................................... 82
5.2 Coarse-grained energy function formulation ............................................ 83
5.3 The Elastic Network Model ...................................................................... 88
5.3.1 Method .............................................................................................. 88
5.3.2 Results ................................................................................................ 92
5.3 Secondary structure formation using continuous optimization ................. 98
5.4 Conformation search using coarse-grained model with rigid secondary
structures .................................................................................................. 102
5.4.1 Method ............................................................................................. 102
5.4.2 Results .............................................................................................. 104
5.5 Discussion ................................................................................................ 110
5.6 Closure ..................................................................................................... 111
6. Simultaneous search in the sequence and conformation spaces:
An application ................................................................................................ 112
6.1 Introduction .............................................................................................. 112
6.2 A brief description the target protein: The hen egg-white Lysozyme ..... 115
6.3 Modeling and results ................................................................................ 117
6.4 Discussion ................................................................................................ 127
6.5 Closure ..................................................................................................... 129
7. Towards parallelization of tertiary structure prediction using Graphics
Processor Unit (GPU) based parallel computation ........................................ 130
7.1 Introduction and motivation ..................................................................... 130

iv
7.2 From CPU-based code to GPU-based code ............................................. 133
7.3 A case study with CPU and GPU based codes ........................................ 136
7.4 Closure ..................................................................................................... 141
8. Closure and future work ................................................................................. 142
8.1 Summary and conclusions ....................................................................... 142
8.2 Contributions of the thesis ....................................................................... 145
8.3 Future work ............................................................................................. 146
Appendix A ......................................................................................................... 149
Appendix B ......................................................................................................... 151
B1 Interior Point Optimization (IPOPT) ....................................................... 151
B2 SCWRL .................................................................................................... 152
B3 Nonlinear conjugate gradient method ..................................................... 152
B4 Online Secondary Structure prediction servers ....................................... 154
Appendix C ......................................................................................................... 156
References .......................................................................................................... 164

v
Abstract
We have developed a novel computational approach to functional de novo protein design
using gradient-based continuous optimization techniques. Motivated by many
engineering optimization applications in which a cost function is optimized subject to a
set of constraints, we pose functional protein design task as a continuous optimization
problem to search sequence and conformation spaces simultaneously. The methods used
in sequence-space search are analogous to the material-design formulations in topology
optimization of structures, whereas the conformation search techniques are similar to
mechanical-link like models and modal analysis of structures. Computationally efficient
techniques such as nonlinear conjugate gradient and interior point optimization are used
to solve the optimization problems. Both the sequence and conformation search
techniques are individually validated with real proteins. Coarse-grained as well as
atomistic level potentials are used to model the energy. Finally, we combined the
sequence and conformation search methods and propose a new strategy for simultaneous
search in the sequence and conformation spaces for designing functionalistic de novo
proteins. In view of lack of experimental resources, the proposed computational scheme
is validated by re-designing an existing protein, the hen-egg white lysozyme. Since the
thrust of this work is on developing computationally efficient models, we developed an
amino acid grouping scheme based on metric multi-dimensional scaling. Some structure-
prediction problems are also solved using Graphical Processing Unit (GPU) based
Compute Unified Device Architecture (CUDA) programming.

vi
Acknowledgments
Pursuing a doctorate degree in an interdisciplinary field at IISc has been the most
memorable achievement in my life. Through this experience I have known myself, my
strengths and drawbacks, and have explored territories that I wouldn’t have even thought
of getting into before I came to IISc. Hence, the largest share of my acknowledgment
goes to this Institute, which has not only molded my way of thinking but also my way of
life, my attitude towards life and society, and my character.
Now, turning to mortal beings, I have to do injustice to so many people by not
acknowledging them directly in this short space, who knowingly or unknowingly, have
helped me through this journey. However, the most prominent one who comes in my
mind is my research supervisor, professor G. K. Ananthasuresh, or Suresh as we call him.
Suresh was my research supervisor in M. Tech., and during this period I was
considerably influenced by his way of teaching, his interdisciplinary topics of research,
and of course his stress on good technical writing and presentations. However, it was
only during PhD that I was able to acquire the skills that are so necessary to convey one’s
ideas and works convincingly in a research paper or a presentation, and for that the credit
goes entirely to Suresh. My PhD topic involved subjects which were new to both of us,
and Suresh was always supportive of the new ideas that I thought of working on. During
the course of my PhD, there were both moments of enjoyment and crisis in my family,
and many times I took leaves which were much longer than what his other students used
to take. I am also grateful to him for allowing me to do so.
Due to the interdisciplinary nature of my work, I had to venture in several
subjects which were new to me. Various courses offered in different departments in IISc
were highly helpful to get initiated into unknown topics. Thus, I am indebted to all the
teachers whose classes I attended. Also, I learned many things relevant to my research
from friends in various departments in IISc, and I feel fortunate to be in such an
academic and research oriented community. I feel especially grateful to Mr. Sumanta
Mukherjee in bioinformatics, and frankly speaking, without his help I might not have
been so successful with the work that I have done. Sumanta helped me in installing the
IPOPT software, which I alone was not able to install in my computer, and further helped

vii
in debugging the C++ codes in which I used to get stuck. He taught me perl and other
scripting languages, which were necessary for running batch computations and parsing
operations, and still a part of my work depends on codes entirely written by him as the
work demanded a high level of codemanship which I have not acquired till now. These
were an invaluable service to an unskilled programmer like me. He was also an eager
helper in my efforts in parallelizing my codes and a part of my parallel codes were
actually tested in his cluster. Regarding parallel programming, I must acknowledge the
help of two of my lab mates, Meenakshi and Ganesh.
Biology was a remote subject to me when I started my work, and my last touch
with biology went back to 10th standard. Sumanta, Amit, Kalidas, Anupam and others
helped me to gain a footing in the area of molecular biophysics which was to be the area
of my research. I am also thankful to my friends Narayana, Sangamesh, Soumyakanti,
Pradipta, Pradeep, Nandkumar, Achintya, Indrajeet, and others who were always
available for a discussion on any theoretical and computational issues. I am especially
grateful to my friends Shamik, Anindya, and Deep, who even though were far away from
IISc, were always in touch with me and supportive of my efforts. And I will always be
thankful to all my friends like Kamalesh, Anirban, Arindam, Ranajit, Subhabrata, Anup,
Satadal, and many others who are like a family to me in IISc.

viii
List of Figures
Figure number Page number
1.1 Analogy between compliant mechanisms and proteins ...………………… 2
1.2 The hierarchic levels of protein structure ………………………………… 5
1.3 Different energy funnels …………………………………………………. 7
1.4 A top view of a four-helix bundle ………………………………………... 8
1.5 The flow diagram of our functional protein design strategy ……………. 15
3.1 Plot of stress against number of dimensions ……………………………. 35
3.2 Scatter diagram showing the discrepancies between entries in the
distance matrix and corresponding distances calculated from the
MMDS map .................................................................................... 36
3.3 MMDS amino acid map constructed using the matrix where we
subtracted diagonal elements from the corresponding rows ....…….. 37
3.4 Amino acid map constructed using the metric multi-dimensional scaling
method and the modified Miyazawa-Jernigan matrix as the proximity
matrix …………………………………………………………………… 37
3.5 Properties of amino acids shown on the MMDS map ………………….. 39
3.6 The residues that have a positive log odd score in the BLOSUM62
matrix are connected by double ended arrows .…………………….. 40
3.7 The residues that have a positive log odd score in the PAM250
matrix are connected by double ended arrows ...…………………… 40
3.8 Dendrogram showing hierarchical grouping of amino acids based
on our distance matrix …………………………………………….. 42
3.9 Minimum distance between groups as a function of the number of
groups …………………………………………………………………… 43
3.10 Grouping of amino acids into five groups based on hierarchical
clustering method…………………………………………............... 44
4.1 Native structure of the four proteins that we target for sequence design.
The number of residues in each protein is also indicated ………………. 46
4.2 In clockwise order from the top ………………………………………… 50

ix
4.3 The double sigmoid function for energy of interaction between all
twenty amino acids …………………………………………………. 52
4.4 Sigmoid function representation of the secondary structure propensities.. 54
4.5 Plot of the constraints. Each colored line is the plot of the constraint ….. 56
4.6 Best designed sequences based on our scoring scheme using
different potentials with and without amino acid composition
constraints for each of the four proteins .………………….. ............. 58-59
4.7 Plots of energy gap ( E native avg decoysE E −∆ = − ) versus Eσ (the standard
deviation of energy of the decoy set structures) …………………….. 63-67
4.8 The highest scoring designed sequences for the four proteins ……… 74-77
4.9 Results of sequence alignment using sequence alignment program
CLUSTAL …………………………………………………………… 77-78
4.10 Plots of energy gap ( E native avg decoysE E −∆ = − ) versus Eσ (the standard
deviation of energy of the decoy set structures) ……………………….. 79
5.1 Variation of contact energy between thi and thj residues ( ijE in kT
units) as a function of distance between them ( ijL in 0A ).…………. 85
5.2 The limits of angle θ between three adjacent Cα atoms ………………. 86
5.3 Variation of bond energy Eθ with angle θ formed by three bonded Cα
atoms ……………………………………………………………………. 87
5.4 Elastic network model of a small de-novo protein, Chignolin ………….. 88
5.5 Flowchart showing our algorithm for large change in conformation
determined using eigenvectors of stiffness matrix K of EN …………… 90
5.6 Energy versus number of iterations for different intervals of
iteration (maxiter) at which optimization program fminunc updates
stiffness matrix of EN ………………………………………………. 91
5.7 Conformation of Chignolin in native state and after optimization.
The left conformation (red lines and blue circles) represent the
native state from PDB…………………………………………......... 92

x
5.8 Fully unfolded conformation of Chignolin……………………………… 93
5.9 The right symmetric half of the energy landscape of Chignolin ………... 94
5.10 (a) The native state of Chignolin (PDB ID 1UAO). (b) Optimal
conformation using last 15 eigenvectors of EN matrix ……………….. 95
5.11 (a) PDB structure of 1GJF. (b) Optimal structure of 1GJF from fully
unfolded state ………………………………………………………….. 96
5.12 (a) Native structure (PDB) of 1RIJ. (b) Optimal structure of 1RIJ from
fully unfolded state …………………………………………………….. 96
5.13 (a) PDB structure of Ubiquitin; the secondary structures have been
shown with bold lines. (b) Conformation after minimization of
energy from native state ….………………………………………. 97
5.14 Schematic diagram of an alpha helix ………………………………….. 99
5.15 OB-CG model of alpha-helix starting from fully unfolded state …….. 101
5.16 The three-dimensional coarse-grained model of a protein. Each
residue is modeled as a bead; the bonds between them are shown
as thick sticks……………………………………………………… 103
5.17 Predicted and native state structures of the protein with PDB ID
1LRE (81 residues)……………………………………………… 105-106
5.18 Predicted and native secondary structures for the proteins 1BCF
(chain A), 1EIJ, 1LYD and 1R69………………………………...... 109
6.1 Schematic of an enzyme molecule. The active site is shown with
bold-dashed lines. Two key residues that form the enzyme substrate
complex are shown with red and blue colored beads .….................... 113
6.2 A ribbon diagram of the hen egg-white lysozyme (PDB ID 1LZE)…… 116
6.3 The GOR4 and HNN servers’ secondary structure prediction results
for the wild-type sequence of 1LZE .......................………………… 119
6.4 Few of the designed sequences having high secondary structure
prediction similarity with wild-type secondary structure of 1LZE... 120-121
6.5 Tertiary structure prediction results using OB-CG model and rigid
secondary structures ....……………………………………………. 122-123

xi
6.6 Plots of energy gap ( E target structure avg decoysE E− −∆ = − ) versus Eσ (the
standard deviation of energy of the decoy set structures) for the
designed sequences..………………………………………………… 126
7.1 The logic of CPU and GPU based codes ……………………………… 132
7.2 Flow diagram of tertiary structure prediction code …………………… 133
7.3 Flowchart of the algorithm evalE_dEdx …………………………. 135-136
7.4 The function value and norm of the gradient of the same for
evalE_dEdx for the CPU and GPU codes ………………............. 139-140

xii
List of Tables
Table number Page number
4.1 Number of matches of the best designed sequences given in Fig.
4.6 using different grouping schemes for each of the four proteins
using all three types of potentials ………………………………….. 60-62
4.2 Average time taken for designing sequences of each protein using
MJ (Miyazawa and Jernigan, 1996), ZSk (Zhang and Skolnick,
1998) and atomistic potentials (Cornell at al., 1995, Fraternali and
Gunsteren, 1996)………………………………………………….... 69
4.3 Average time taken to design sequences for each protein in the FISA
decoy set using quadratic programming formulation ………………….. 72
5.1 Table showing number of iterations required by the optimization
program fminunc to converge as the stiffness matrix of K is
updated after a particular number of iterations for Chignolin …….. 91
5.2 Protein structure prediction results using nonlinear conjugate
gradient algorithm. The first column shows the PDB id. of the
protein, the number of residues in it, and the percentage of residues
in secondary structures (α -helices)……………………………..... 107-108
5.3 Results of ab initio structure prediction with secondary structures
predicted by the HNN server ..…………………………………... 110
6.1 Few selected examples of tertiary structure prediction results using
both energy models. Under each energy model, the first column
indicates the DRMSD of the unfolded conformation from 1LZE
(C α− coordinates only) which serves as input to optimization
program……………………………………………………….…... 124
7.1 Time required for different calculations in the function
evalE_dEdx in CPU and GPU. The calculations are named
similarly as they are presented in Fig. 7.3. ……………………....... 137

xiii
List of Equations
Equation number Page number
1.1…………………………………………………………………………….. 14
1.2…………………………………………………………………………….. 16
3.1…………………………………………………………………………….. 34
3.2…………………………………………………………………………….. 34
3.3…………………………………………………………………………….. 34
3.4…………………………………………………………………………….. 35
3.5…………………………………………………………………………….. 35
4.1…………………………………………………………………………….. 50
4.2…………………………………………………………………………….. 51
4.3…………………………………………………………………………….. 51
4.4…………………………………………………………………………….. 51
4.5…………………………………………………………………………….. 51
4.6…………………………………………………………………………….. 53
4.7…………………………………………………………………………….. 53
4.8…………………………………………………………………………….. 53
4.9…………………………………………………………………………….. 53
4.10…………………………………………………………………………… 53
4.11…………………………………………………………………………… 55
4.12…………………………………………………………………………… 55
4.13…………………………………………………………………………… 62
4.14…………………………………………………………………………… 69
4.15…………………………………………………………………………… 69
4.16…………………………………………………………………………… 70
4.17…………………………………………………………………………… 70
4.18…………………………………………………………………………… 71
4.19…………………………………………………………………………… 71
4.20…………………………………………………………………………… 78
5.1…………………………………………………………………………….. 84

xiv
5.2…………………………………………………………………………….. 85
5.3…………………………………………………………………………….. 86
5.4…………………………………………………………………………….. 87
5.5…………………………………………………………………………….. 87
5.6…………………………………………………………………………….. 89
5.7…………………………………………………………………………….. 99
5.8…………………………………………………………………………… 102
5.9…………………………………………………………………………… 103
5.10………………………………………………………………………….. 104
6.1…………………………………………………………………………… 118
6.2…………………………………………………………………………… 118

1
1. Introduction
• A preamble to the thesis is given.
• Brief reviews of protein structure and folding are presented.
• Brief review of protein design is given.
• The motivation for the work is described.
• Protein design is posed as an optimization problem.
• The scope of the thesis is noted.
• The organization of the thesis is described.
• The chapter is closed with a brief summary.
1.1 Preamble
This thesis presents work on computational design of protein molecules for structural and
functional specifications using gradient-based optimization. Proteins are molecular
machines that perform life-sustaining functions, for example, decoding genetic
information, catalyzing bio-chemical reactions, triggering immune response, sustaining
rigidity and shape of cells and tissues, facilitating chemical signaling among cells, etc.
(Brandon and Tooze, 2001). The sequence of amino acids along a protein’s linear chain
determines its folded structure, also called the conformation, which is crucial to its
specific function. Thus, the protein design problem entails the determination of the amino
acid sequence so that it folds into a suitable 3D structure to serve a desired function.
Optimization is inherent in protein design because a protein chain folds into a native
conformation that, reportedly, has the minimum free energy (Anfinsen, 1961, 1973) with
respect to other conformations.
This work is motivated by the broad principles that underlie optimal design of
machines and structures, and compliant mechanisms in particular. Compliant
mechanisms are elastically deformable structures (Howell, 2001). Figure 1 depicts the
analogy between proteins and compliant mechanisms. Both need specific structural forms
to perform their function and change their shape to do it. Just as a protein’s structure and
function are determined by its sequence of amino acids, a compliant mechanism’s
function is decided by its geometry and material. The deformed configuration of a

2
compliant mechanism is governed by the principle of minimum potential energy
analogous to the principle of minimum free energy of a protein obeys while folding. By
Fig. 1.1 Analogy between compliant mechanisms and proteins.
a) A compliant mechanism (a gripper) in the open position.
b) The same gripper in the closed position.
c) A protein (hexokinase) in its non-active (open) position (Adapted from Nelson
and Cox, 2008).
d) The same protein in its active (closed) position. The active site is encircled in the
figure(Adapted from Nelson and Cox, 2008).
(c)
Active site
(d)
(a)
(b)

3
taking advantage of the analogy between proteins and compliant mechanisms and
computationally efficient optimal design techniques developed for compliant mechanisms
and mechanical structures, this thesis adopts a new approach to computational protein
design. We pose de novo protein design (i.e., designing a protein anew) as an
optimization problem wherein the site of action of the protein is specified in terms of its
structure and amino acids as illustrated in Fig. 1.1.
The aspects of protein design considered in the thesis include: (i) reduced amino
acid alphabet that simplifies protein sequences, (ii) search in the sequence space using
continuous modeling, (iii) search in structure space using coarse-grained energy
potentials, and (iv) simultaneous search in sequence and structure spaces using coarse-
grained potentials as well as fine-grained atomistic potentials. While the design
philosophy of the thesis is general and independent of the potentials, we do consider
instances of real proteins to illustrate the efficacy of the proposed methodology.
Before explaining the specific motivation and the scope of the thesis, requisite
background to the different aspects of the work is provided next.
1.2 Proteins
1.2.1 A brief overview of protein structure and folding
Proteins are biopolymer chains made of monomers called amino acid residues (see
Appendix A). They constitute an important class of biomolecules which take part in all
life-sustaining processes. Proteins are the most versatile biomolecules in terms of the
functions they perform. A few activities in which proteins take active part are:
deoxyribonucleic acid (DNA) duplication, DNA to ribonucleic acid (RNA) transcription,
mediating biomolecular reactions, biosignalling, cytoskeleton generation, bioenergetics,
etc. The diverse functions that proteins are able to perform are due to their structure, i.e.,
spatial conformation. This has been possible because proteins differ from other
biopolymers in one significant aspect; unlike other polymers whose molecules exist in
randomly coiled (glassy) state under normal conditions of temperature, chemical and
other environmental conditions (such as those that exist on our planet), molecules of a
particular protein under most of these conditions have a remarkable similarity in
structure. Thus, all molecules of hemoglobin in our red blood cells have a particular

4
structure when they are transporting oxygen, and a slightly different structure when they
are transporting carbon-dioxide.
The protein structure is hierarchic, with three to four levels of hierarchy clearly
identifiable in most protein structures (Nelson and Cox, 2008). The first level, known as
the primary structure or the sequence of the protein, comprises the order of the amino
acid residues in the polymeric chain of the protein (Fig. 1.2 a). At this level there is no
geometrical information conveyed in the structure. In the next level, local geometrical
patterns form on the polymeric chain of the protein (called the backbone) aided by the
formation of hydrogen bonds and constraints in the free movement of the backbone
(called the steric constraints). These are known as secondary structures, and are
classified according to the geometric shapes they most closely resemble:, helix, sheet and
turn. The most widely occurring secondary structures are the alpha (α ) helices and the
beta ( β ) sheets (Fig. 1.2 b). The secondary structures are closely packed to form the next
higher level structure like a globule or channel, known as the tertiary structure of the
protein (Fig. 1.2 c). The formation of a tertiary structure is governed by a complex
interplay of molecular forces. Sometimes, a protein may consist of more than one chain
that assemble together to form a large complex structure, known as the quaternary
structure (Fig. 1.2 d).
New polypeptides are synthesized inside the cell in an organelle called the
ribosome. As the newly synthesized polypeptide emerges from the ribosome, it rapidly
folds (in the order of micro to mille seconds) to a characteristic three-dimensional
structure, called the native structure of the protein. The rapid folding of the polypeptide is
governed by the minimization of its free energy (Anfinsen, 1961, 1973, Onuchic et al.,
1997). How a large molecule like protein with high number of degrees of freedom can
rapidly find a stable conformation is often expressed in terms of what is known as
“Levinthal’s paradox” (Levinthal, 1968). Proteins fold under the action of a number of
forces, namely, hydrophibic-hydrophilic interaction among side chains, hydrophilic
interaction with water, hydrogen bonding within the backbone (α -helix and β -sheet
formation) and with surrounding water, ionic interactions among polar residues (salt
bridges), di-sulphide bond formation, vander Waals forces and electrostatics.
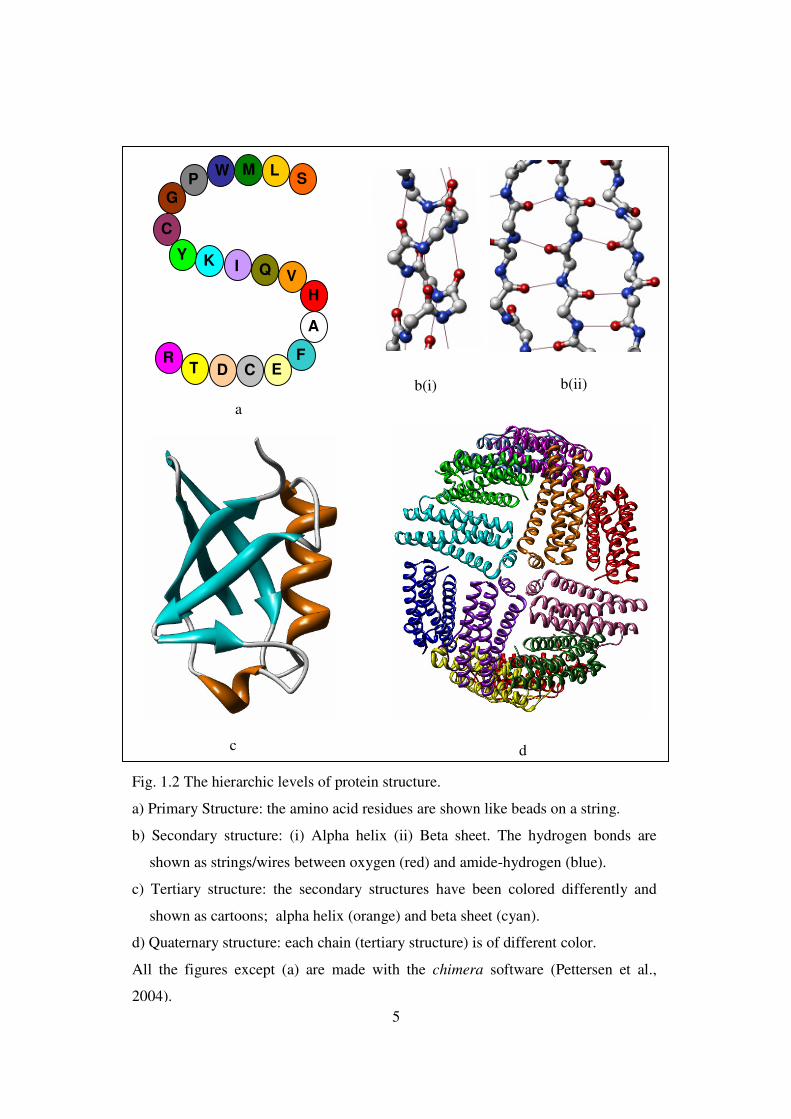
5
b(i) b(ii)
c d
Fig. 1.2 The hierarchic levels of protein structure.
a) Primary Structure: the amino acid residues are shown like beads on a string.
b) Secondary structure: (i) Alpha helix (ii) Beta sheet. The hydrogen bonds are
shown as strings/wires between oxygen (red) and amide-hydrogen (blue).
c) Tertiary structure: the secondary structures have been colored differently and
shown as cartoons; alpha helix (orange) and beta sheet (cyan).
d) Quaternary structure: each chain (tertiary structure) is of different color.
All the figures except (a) are made with the chimera software (Pettersen et al.,
2004).
M
G P
W L
I
A
T C
C
F
V
Y
R
H
Q
S
D E
K
a

6
However, recent views substantiated by atomic level experiments and extensive computer
simulations hold that the favorable increase in entropy, which occurs when hydrophobic
residues are packed in the interior of the protein starts the initial folding process (known
as the “hydrophobic collapse”); subsequently the initial folded state, also known as the
“molten globule” is stabilized by the formation of secondary structures, di-sulphide bonds
and ionic interactions among polar residues (Dill, 1990, Nelson and Cox, 2008). The
recent view of protein folding is explained in terms of “the energy landscape” or the
“folding funnel” (Wolynes, 2004). “The new perspective sees folding as a diffusion-like
process, where the motions of individual chains are asynchronous, each being buffeted by
Brownian forces through different sequences of chain conformations, which ultimately
all find their ways to the same native structure, in the same way that water flowing along
different routes down mountainsides can ultimately reach the same lake at the
bottom…..Since the lateral area of an energy landscape at a given depth represents the
number of conformations having the given intra-chain free energy, the funnel idea is
simply that as a folding chain progresses towards lower intra-chain free energies—by
increasing compactness, hydrophobic core development, intra-chain hydrogen bonding,
salt-bridge formation, and so forth—the chain’s conformational options become
increasingly narrowed, ultimately towards one native structure.” (Dill and Chan, 1997).
The different energy landscapes for explaining different observations of protein folding
have been shown in Fig. 1.3. Even though the theoretical framework of protein folding
has been satisfactorily explained based on energy-landscapes, computationally folding a
polypeptide from the conformation when it is released from the ribosome to the native
state is still a daunting task.
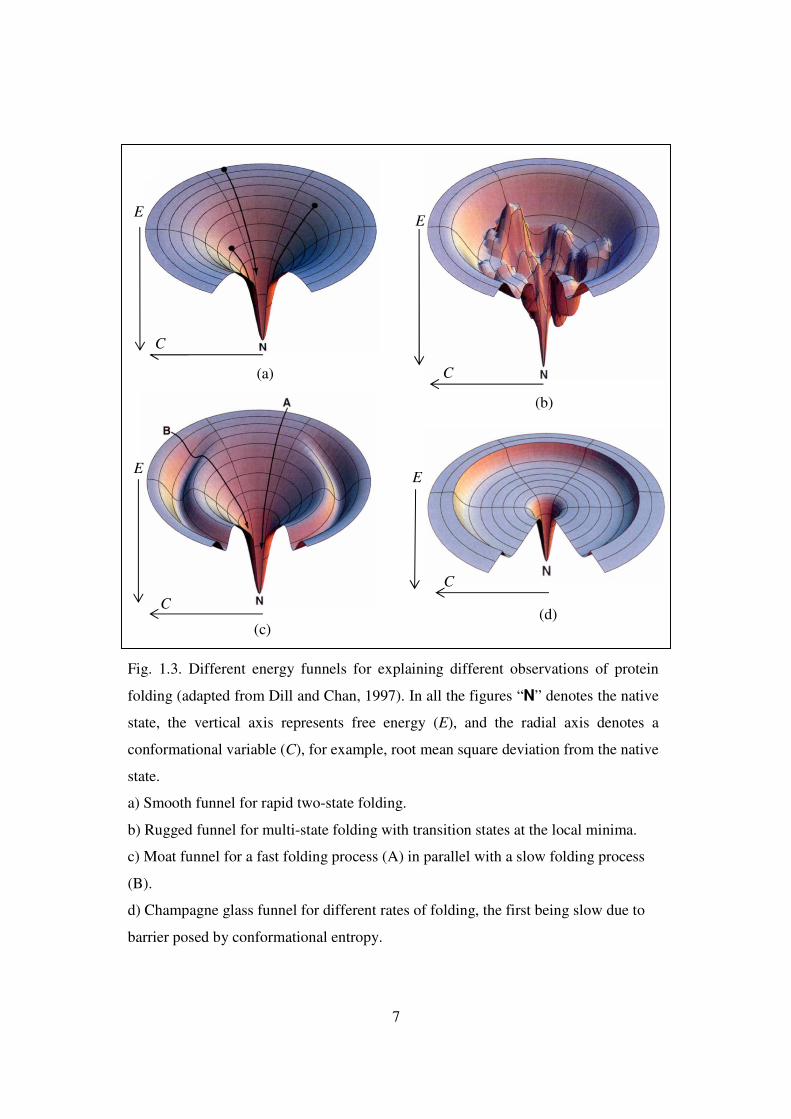
7
Fig. 1.3. Different energy funnels for explaining different observations of protein
folding (adapted from Dill and Chan, 1997). In all the figures “N” denotes the native
state, the vertical axis represents free energy (E), and the radial axis denotes a
conformational variable (C), for example, root mean square deviation from the native
state.
a) Smooth funnel for rapid two-state folding.
b) Rugged funnel for multi-state folding with transition states at the local minima.
c) Moat funnel for a fast folding process (A) in parallel with a slow folding process
(B).
d) Champagne glass funnel for different rates of folding, the first being slow due to
barrier posed by conformational entropy.
(a)
E
C
(b)
E
C
(c)
E
C (d)
E
C

8
1.2.2 A brief overview of protein design
There are two goals of protein design. The first is to design proteins from the first
principles, or de novo design as it is known, with an aim to understand the underlying
physical principles that govern protein folding (DeGrado et al. 1991). The goal in this is
to design amino acid sequences that will adopt a “unique and stable three-dimensional
structure” (Yue and Dill 1992). The second goal is to “create proteins with desired
functions” (Pokala and Handel 2001).
Fig. 1.4. A top view of a four-helix bundle. The helices are represented by helix
wheel representation using a repeat of 3.6 residues per turn. The polar residues are
shown as white circles and non-polar residues as black circles around the helix
wheel. It can appreciated from this figure that the core of the four-helix bundle is
composed of hydrophobic residues buried inside the protein (Adapted from
Kamtekar et al, 1993).

9
The first attempts of de novo protein designers were secondary structures such as helices
and strands, which under the action of hydrophobic forces self-assemble to form globular
protein-like conformations (Sym et al., 1984, Ho and DeGrado, 1987, Chin et al., 1992).
The design of self-assembling secondary structures was followed by the de novo design
and creation of coiled coils (Hodges et al., 1990, Cohen and Parry, 1990) and four-helix
bundles (Regan and DeGrado, 1988, Hecht et al., 1990, Kamtekar et al., 1993,
Schafmeister et al., 1997), which are among the simplest of all helical proteins observed
in nature. There have been attempts to design β -sheet proteins, but these designs were
not as successful as those of the α -helical bundles (Yan and Erickson, 1994, Hecht,
1994, Quinn et al., 1994). The successful design of helical bundles prompted designers to
formulate simple heuristic rules (Hecht, 1994, DeGrado, 1999); for example, “binary
patterning” of hydrophobic and hydrophilic residues for making the core of the designed
proteins (Kamtekar et al., 1993, Hecht, 1996, Woolfson, 2001, Ventura and Serrano,
2004). In binary patterning, the polar and non-polar residues are positioned on the
secondary structures periodically such that the secondary structures attract one another
and form a hydrophobic core like that of a globular protein (see Fig. 1.4). The design
procedure of such de novo proteins is described in detail in a few reviews (DeGrado,
1988, Sander, 1994, Gibney et al., 1997, Schafmeister et al., 1998).
The design of helix-bundles by simple heuristic rules is possible because of their
topological simplicity. However, this is not true for the de novo design of globular
proteins in general (Woolfson, 2001). De novo sequence design is a computationally
challenging task, which is argued to be an NP-hard problem (Pierce and Winfree, 2002).
The computational algorithms that are widely used for de novo sequence design can
be divided into two broad categories: combinatorial and heuristic (Desjarlais and Clarke,
1998). The combinatorial or the pruning approach, first simplifies the search space by
allowing certain discrete conformations. Then, by systematically applying a rejection
criterion, a number of the combinatorial possibilities are eliminated (Desmet et al., 1992,
Gordon and Mayo, 1999). The advantages of these algorithms are that they are robust and
can search a function for a global minimum, provided it exists. The problem of
combinatorial algorithms is that they become computationally expensive as the sequence
size grows (Voigt et al., 2000) or if the flexibility of the backbone is to be incorporated;

10
in the latter case heuristic rules have been applied (Harbury et al., 1998, Wernisch et al.,
2000). The second class of algorithms search the sequence space in a semi-random
manner that depends both on the energy landscape and algorithm-specific rules. The most
widely used algorithms of this type are the Monte-Carlo (MC) method (Metropolis et al.,
1953, Lee and Levitt, 1991, Hellinga and Richards, 1994, Dahiyat et al., 1997, Irbäck et
al., 1998, Kuhlman et al., 2003) and genetic algorithms (GA) (Holland, 1992, Tuffery et
al., 1991, Desjarlais and Handel, 1995, Pedersen and Moult, 1996, Raha et al., 2000). The
advantage of these algorithms is that they can be applied for sampling energy functions
and conformational spaces which are much more complicated than those handled by
combinatorial techniques; in particular, rotamer and backbone conformations can be
varied continuously (Hellinga and Richards, 1994, Desjarlais and Handel, 1999).
However, there is no guarantee that these algorithms will converge to a global minimum
(Desjarlais and Clarke, 1998, Voigt et al., 2000), or worse, they may converge to
different solutions depending upon different parameters used in the program (Goffe et al.,
1994). More recently, mean field theory-based approaches are used to identify the most
probable set of sequences for a given structure (Saven and Wolynes, 1997, Zou and
Saven, 2000, Kono and Saven, 2001). However, such techniques use statistically derived
potentials which may not have a physically realistic basis, and hence, are biased to the
particular set of structures for which the mean field is derived (Thomas and Dill, 1996,
Moult, 1997, Zhang and Skolnick, 1998).
The main goal for the development of de novo protein design computation techniques
is to help the experimental researchers in creating de novo proteins. To this end, a few of
the abovementioned algorithms have successfully helped researchers in making
sequences that have folded to correct target structures (Dahiyat and Mayo, 1997a,
Harbury et al., 1998, Bryson et al., 1998, Kraemer-Pecore et al., 2001, Kuhlman et al.,
2003).
Let us now turn to the second goal of protein design, i.e., design of proteins with
desired functions.
De novo protein designers have been successful in altering the activities/specificities
of some natural proteins by slightly modifying their sequences. These include: alteration
of DNA-binding specificity (Wharton and Ptashne, 1985), alteration of cofactor

11
specificity (Scrutton et al. 1990), alteration of substrate specificity (Hedstrom et al.,
1992), metal binding activity (Kuroki et al., 1989, Hellinga et al., 1991, Inaka et al.,
1991), site-specific-DNA-cleavage (Sluka et al., 1987), design of catalytic antibodies
(Lerner et al., 1991), etc. The design of novel proteins capable of binding to specific
ligands was achieved as early as 1979 by Gutte and co-workers (Gutte et al., 1979,
Jaenicke et al., 1980, Moser et al., 1983, Klauser et al., 1991). Considerable success is
achieved in the design of metal-binding proteins (for a detailed review, see DeGrado et
al., 1999). Membrane proteins are critical to many biological processes, and the design of
de novo membrane proteins with tailor-made activities is a significant step towards
achieving the aforementioned second goal of de novo protein design (Montal et al., 1990,
Oiki et al., 1990, DeGrado and Lear, 1990, Grove et al., 1991). In this vein, it is worth
noting that protein-like polymeric materials are developed to structurally change (expand
or contract) in response to changes in temperature, pH, etc. (Urry, 1990, Luan et al.,
1991); further, protein-like modules that self-assemble into hollow nanotubes are also
reported (Ghadiri et al., 1993).
The computational design of proteins with de novo functions pose significant
challenges to researchers in the pertinent field. The incorporation of functional specificity
entails considerable conformational flexibility of the backbone (Lassila, 2010). However,
with increase in the backbone flexibility, computational cost increases exponentially
because of exponential rise in the number of allowable rotamer states and corresponding
energy calculations. Recently, aided by high computational power and efficient
techniques, researchers were able to design a few proteins with novel functions (Bolon
and Mayo, 2001, Looger et al., 2003, Dwyer et al., 2004, Jiang et al., 2008, Rothlisberger
et al., 2008, Siegel et al., 2010). However, the performance of functionally designed de
novo proteins (say, enzymes) compared to their natural counterparts has raised sensitive
questions about the efficacy of the present theory underlying the computational methods
(Baker, 2010).
The present computational scenario for functionally active de novo protein design
provides a suitable background for the motivation of the work presented in this thesis.

12
1.3 Motivation
The preceding section gave a brief overview of computational protein design. Most of the
current computational methods for de novo sequence design are exclusively designed for
a fixed backbone structure with a few notable exceptions that allow for perturbations (Su
and Mayo, 1997, Harbury et al., 1998, Desjarlais and Handel, 1999, Kuhlman et al.,
2003). However, a true protein design strategy requires simulation and search in both
sequence and structure spaces (Schueler-Furman, 2005). Perhaps because of limited
computation power, until recently simultaneous searching in both sequence and structure
spaces was a difficult task for computational scientists. However, as de novo protein
design is entering a new era of functional de novo protein design, it is clear that
computational scientists have to design methods to efficiently search the sequence and
conformation spaces simultaneously (Mandell and Kortemme, 2009, Baker, 2010). We
are motivated by this requirement, and, in this thesis, present a novel approach for
efficient search of sequence and conformation spaces simultaneously with a view to
design proteins with predefined functions.
We formulate de novo protein design with predefined functions as a classic
constrained optimization problem consisting an optimization function of several variables
obeying a set of constraints. The general nature of such an optimization problem is shown
in Eq. (1.1). As we pose the problem in terms of continuously differentiable mathematical
functions we are in a position to utilize the mathematical framework of optimization
theory, the necessary and sufficient conditions for determining a local optimum, and the
Karush-Kuhn-Tucker conditions for determining Lagrange multipliers to solve nonlinear
optimization problems with continuously differentiable constraints (Luenberger,
Papalambros and Wilde, 2000). With the mathematical framework of the optimization
theory as our base, we use gradient-based optimization algorithms, for example,
conjugate gradient (CG) and sequential quadratic programming (SQP) to solve the
optimization problem. Gradient-based continuous optimization algorithms are efficient in
determining local minimum deterministically (with or without constraints); some of these
algorithms can solve a convex problem of n variables in O(n) steps (Luenberger,
Shewchuk 1994).

13
It should be noted from the preceding section that the computational algorithms for
protein sequence design, which are highlighted in the relevant literature, are mostly
combinatorial or heuristic as can be discerned from the review papers on de novo protein
design (see Chapre2)). Even in the case of protein structure prediction, the algorithms
most widely used are either molecular dynamics (Levitt, 1983, Case et al., 2005, Hess et
al., 2008) or Monte Carlo (Das and Baker, 2008) or heuristic techniques based on
template-matching (Sali and Blundell, 1993). However, we have chosen to use gradient-
based continuous optimization algorithms both for de novo protein sequence design and
for protein structure prediction. It can also be noticed from the brief overview of the
literature presented in the preceding section (and in Chapter 2 in detail) that much
emphasis is placed on discrete rotamer states for energy calculations. This often becomes
a bottleneck in terms of computation power, speed, and computer memory. However,
when there are relatively large changes in conformational states compared to side chain
movements, as it happens in case of an enzyme or a ligand binding protein, coarse-
grained structure prediction present an efficient way of conformational sampling than
their fine-grained counterparts (Mandell and Kortemme, 2009). We, in our approach use
coarse-grained structure prediction as it presents a way of searching a space that is almost
infinite1 at low computational cost. By combining our gradient-based optimization
programs for designing sequences and searching the conformation space, we present a
novel strategy for simultaneous search in sequence and conformation spaces, which is
described in the following section.
Before moving on to the next section, it is useful to mention a few things, which
might help the reader understand the philosophy of this work. Our endeavors to propose
novel formulations or use novel techniques with a view of computational efficiency have
sometimes led us to develop new methods, which, at the first sight, may appear unrelated
to the overall goal of functional protein design. We developed an amino acid grouping
scheme using metric multidimensional scaling for grouping amino acids with a view to
work with a reduced amino acid set for sequence design (Rakshit and Ananthasuresh,
2008), but later we used robust gradient-based optimization algorithms that can solve the
1 A large molecule such as a protein has very many number of degrees of freedom even after satisfying the constraints of the Ramachandran map, a fact that Levinthal used to pose an eponymous paradox.

14
sequence design problem with the full set of twenty amino acids. We also developed an
elastic network (EN) approach for tertiary structure prediction (Rakshit and
Ananthasuresh, 2010) with the goal of working with fewer variables than the full set of
residue coordinates by using the mode shapes of EN. However, we found that it was not
so as the calculation of mode shape itself proved to be an additional burden for efficient
computation. Subsequently, we did not follow this approach for tertiary structure
prediction. Thus, this work should not be judged as one which is well rounded-up and
finished; but rather as start of a new approach to computational protein design that is
complementary to approaches pursued by mostly biology researchers for over half a
century.
1.4 Problem Statement
Optimization problems can be broadly formulated as follows:
/ :
design variables
Objective FunctionMinimize Maximize
Subject to :
Governing Principle
Constraints
(1.1)
The governing principle that guides a protein molecule to fold it to its native structure
among a myriad of other possible structures is the minimization of its free energy
(Anfinsen 1961, 1973). We consider the minimization of free energy of the protein
molecule as the objective function in our protein design problem. We have two types of
design variables, material { }ρ and geometric { }x . The material variables are the types of
residues at a particular position in the sequence of the protein, whereas the geometric
design variables are the quantifiers for the position of the residues in space. Thus, while
designing the protein, one has to minimize the free energy in both the sequence (material)
and structure (geometric) spaces. The functional requirements of the protein (i.e., the
particular type of residue which takes part in a reaction or where ligand-binding takes
place) as well as the geometrical requirements of the structure (for example, the geometry
of the binding site in case of an enzyme (see Fig. 1.1) can be specified as constraints in
the optimization problem). One may also impose constraints on the composition of amino
acids, i.e., number of each type of amino acid, which the designed protein ought to have.

15
iii) Generate an ensemble of native-like tertiary structures.
vi) Test the sequences for specificity to the tertiary structure given by (iv) with respect to the ensemble of tertiary structures generated in (iii). vii) Select sequences with high Z-score.
i) Given the specified structure and residues, design the best possible sequences.
ii) From the designed sequences, predict secondary structures (alpha helices and beta sheets).
iv) Select the tertiary structure closest to the target structure based on a suitable metric.
v) Design sequences based on the tertiary structure given by (iv).
Fig. 1.5. The flow diagram of our functional protein design strategy.

16
Such a compositional constraint is necessary for sequence-structure specificity
(Shakhnovich and Gutin, 1993, Koehl and Levitt, 1999). Based on the abovementioned
design criterion and constraints the functional protein design problem is be posed as
( )
�{ } { } �
�{ } { } �
{ } { }
1
0
0
: ,
: : ; 1
: : ; 1
: ; 1 , 1 20
,
j
k
N
i i
i
j
k
ij j i
Minimize E x
Subject to
Material constraints j N
Geometric constraints x x x x k N
Composition constraints M n j N i
x
ρ
ρ ρ ρ ρ
ρ
ρ
=
∆
⊂ = ≤ <
⊂ = ≤ <
≤ ≤ ≤ ≤
∈ ∈
∑
=
� �
(1.2)
The step-by-step approach of our design strategy is as follows.
i) Given the specified structure and residues, design the best possible sequences.
ii) From the designed sequences, predict the secondary structures (alpha helices
and beta sheets).
iii) Select sequences with predicted satisfactory secondary structures and generate
an ensemble of energy minimized tertiary structures.
iv) Select the tertiary structure closest to the target structure based on a suitable
metric.
v) Design sequences based on the tertiary structure given by (iv).
vi) Test sequences for specificity to the tertiary structure given by (iv) with
respect to the ensemble of tertiary structures generated in (iii).
vii) Select sequences with high Z-score.
viii) Go to (ii) and iterate.
The flow diagram of this design strategy is presented in Fig. 1.5. Although we have
chosen to use continuous optimization as our main computational tool, we were not able
to solve all the steps of the abovementioned design strategy using continuous
optimization algorithms. The limitations and the scope of this work are presented in the
following section.

17
1.4 Scope of the thesis
We now outline the scope of the work described in this thesis. Our functional protein
design method is suitable for single-domain proteins, although, we believe that designing
multi-domain proteins will be an extension of our approach. Further, the number of
residues is to be specified a priori. If the numbers of amino acids of each type are
specified, then by imposing constraints on amino acid composition, specificity conditions
can be ensured during sequence design. The identification of the residues and the part of
the structure which forms the basis for the functional activity of the designed protein, is
also to be specified a priori, and should not be changed during the iterative design
process.
We have tried, to the best of our efforts, to adhere to continuous optimization
algorithms, but in certain cases, it was not possible. For example, secondary structure
prediction could not be formulated as a mathematical problem involving continuous
mathematical functions. In such cases, we have used freely available tools, for example,
web-based secondary structure prediction servers and programs for optimal side-chain
packing. Consequently, our results are be limited by the effectiveness of such tools.
Furthermore, we have used coarse-grained energy models to predict tertiary structures.
The issue of using coarse-grained (CG) models for predicting protein structures is an
often a topic of debate (Tozzini, 2010), for example, the applicability of coarse-grained
models to predict the formation of secondary structures (Sancho and Rey, 2006), or
inability of CG models to predict disulphide bonds. However, in this work we were more
concerned with searching the conformation space of a designed sequence about whose
structure nothing is assumed (except the small part which is specified as a constraint in
the problem). Hence, for computational efficiency, we chose to use CG models (C-α
atoms) with the view that the best candidate CG structures for the structure of the de novo
protein determined from our simulations can be supplemented by fine-tuned simulations
such as molecular dynamics. We have used a few CG energy models, namely, Miyazawa-
Jernigan (MJ) matrix (Miyazawa and Jernigan, 1996), Zhang and Skolnick matrix (Zhang
and Skolnick, 1998) and Levitt’s coarse-grained potentials (Levitt, 1976) for our tertiary
structure prediction program. We do not question the applicability of these CG energy
models in our functional de novo protein design strategy; rather we assume that they are

18
applicable. Hence, the results presented here will also be limited by the efficacy of these
potentials and energy matrices.
In summary, the spirit of this work is to be understood as an effort to treat protein
design problem differently from the existing approaches. The underlying philosophy is to
develop techniques that are amenable for gradient-based optimization techniques that are
known to be computationally efficient. The wherewithal needed to do this depends on the
concepts and techniques developed in the pertinent fields. Therefore, the methodology
presented in this thesis will come to fruition as all the related aspects also reach a state of
maturity and general acceptance. Nevertheless, efforts are made in this work to present
practicable results to the extent possible. Numerous examples, some realistic and
biologically relevant, are included.
1.6 Organization of the thesis
This thesis is organized into five broad divisions depending upon their individual
objectives. The first one is the introduction (Chapter 1, this chapter), the main purpose of
which is to introduce the subject matter of this work and explain the motivation. This is
followed by literature review (Chapter 2) on different computational methods that are
related to the methods we have used or developed as also the ultimate goal that we have
in view. After that, we describe the methods that we have developed keeping in mind the
ultimate goal of functional de novo protein design. The first method that we developed
was grouping of amino acids into a reduced alphabet set (Chapter 3). This is followed by
de novo design of sequences for fixed backbones, in which we present two novel methods
using continuous optimization (Chapter 4). As our work requires us to perform search
both in sequence and conformations spaces, we have also developed our method of
predicting protein tertiary structures using coarse-grained models and continuous
optimization. In the case of tertiary structure prediction, we developed two methods the
first of which uses elastic networks and the second, mechanistic linkage models (Chapter
5). We give a brief description of the progress from the prediction of primary structure
(the sequence) to the tertiary structure through an intermediate step of prediction of
secondary structures using secondary structure prediction servers. Finally, we combine all
our methods into the goal of designing a protein with predefined structural and functional
constraints (Chapter 6). We present the results in light of the strategy outlined in the

19
introduction. A brief section describing our efforts towards parallelizing our structure
prediction computer code using Graphics Processing Unit (GPU) based Compute Unified
Device Architecture (CUDA) technology is also included (Chapter 7). We end the thesis
with a concluding section where we discuss how this work may be extended in future
(Chapter 8).
1.5 Closure
We conclude this chapter by briefly summarizing what we have discussed till now. At the
outset, we explained the subject matter of this thesis and gave a brief overview of two
topics related to this work, namely, protein structure and folding, and de novo protein
design. Then, in the light of the emerging trends in de novo protein design, namely,
design of functionalistic proteins, we explained the motivation of this work. Next, we
presented the formulations of functionalistic de novo protein design as a continuous
optimization problem obeying a governing principle and subjected to a set of constraints.
We also gave a broad overview of our design strategy to help the reader in viewing our
computational strategy for functionalistic de novo protein. This was followed by the
scope of the work presented in this thesis. Finally, we described the organization of this
thesis before closing this chapter.

20
2. Literature Review
• We present a literature survey on the different techniques of simplifying the
amino acid alphabet set.
• We review the computational techniques on protein sequence design.
• We present a literature survey on elastic networks and its applications.
• We present a review on minimalist coarse-grained models pertaining to our work.
• The chapter is closed with a brief summary.
2.1 Reduced amino acid alphabet
The folding of a protein is governed by the information stored in its amino acid sequence
(Anfinsen, 1961, 1973). Amino acids, which are 20 in number, can be broadly classified
as hydrophobic and hydrophilic (Nelson and Cox, 2008). Hydrophobic collapse is one of
the dominant forces that govern folding of globular proteins (Chotia, 1984, Dill, 1990).
This notwithstanding, a broad classification into only two categories is often not
sufficient for better understanding of the evolution of proteins, conservation of protein
structures when some amino acids are substituted by others, and the general principles
underlying protein folding and design (Wolynes, 1997). Thus, grouping the amino acids
into simplified sets of more than two seems beneficial.
Dayhoff and co-workers (1972) were the first to quantify the relation between
amino acids by calculating the Relatedness Odds Matrix based on the common ancestry
of proteins. They classified the amino acid residues into five sets based on the chemical
properties of the residues. Based on the work of Dayhoff et al., French and Robson
(1983) used multidimensional scaling (Kruskal, 1964) to elucidate the gradual variation
of hydrophobicity when plotted on a two-dimensional map. Subsequently, with the
availability of a large number of experimentally solved protein structures and with the
high number of protein sequences to be threaded to these structures for suitable structure-
sequence matches, a number of reduced amino acid sets have been deduced based on
different criteria and different computational methods. A brief overview of such methods
follow.

21
Wang and Wang (1999) did an exhaustive enumeration of the “mismatch”
between different amino acids to put forward different reduced sets of simplified amino
acid alphabet varying from two to twenty. Their work was based on the Miyazawa
Jernigan (MJ) matrix (1996). They noted that the best number of reduced alphabets is
five, and they claimed it to be in agreement with the experimental work of Baker’s group
(1997). In a more recent work, Wang and Wang (2002) noted that there is a saturation
with respect to mismatches when the number of the simplified sets is around 10. Li et al.
(1997) did eigenvalue decomposition of the MJ matrix and came to the conclusion that
the MJ matrix reflected interaction of two main forces in protein folding, namely, the
hydrophobic force and the force of demixing that obeys Hildebrand’s solubility theory of
simple liquids.
Murphy et al. (2000) proposed a hierarchic grouping of the amino acids based on
correlation coefficients deduced from the BLOSUM 50 matrix (Heinkoff and Heinkoff,
1992). Cieplak et al. (2001) also did eigenanalysis of the MJ matrix by considering the
“distances” between the amino acids and classified them into five groups. Venkatarajan
and Braunn (2001) used principal component analysis (Johnson and Wichern, 2006) for
creating amino acid maps using large data sets. They used 237 physical-chemical
properties of amino acids to form a vector in a 237-dimensional space for each amino
acid and reduced the resulting matrix to a five dimensional space by using the first five
eigenvalues and eigenvectors. Cannata et al. (2002) applied the branch and bound
algorithm to evaluate all possible groupings of the amino acids based on the PAM
(Schwartz and Dayhoff, 1978) and BLOSUM (Heinkoff and Heinkoff, 1992) matrices. Li
et al. (2003) devised a global alignment method based on substitution matrices and
similarity scores and used the Monte Carlo algorithm to arrive at a reduced set for the
amino acids. Koisol et al. (2003) introduced a Markovian model of grouping the amino
acids that depends on amino acid replacement rate as proteins undergo mutation in
evolution.
More recently, Luthra et al. (2007) used the method of multidimensional scaling
(Kruskal, 1978) to calculate the inter-residue potentials of five reduced groups of amino
acids based on the MJ matrix. Rakshit and Ananthasuresh (2008) also used metric
multidimensional scaling to construct low-dimensional maps of amino acids based on the

22
MJ matrix. They showed that when the amino acids are plotted as points on a two-
dimensional map, there is a directional increase of hydrophobicity from one end to the
other. Based on their analysis, they concluded that the best representative number of
reduced amino acid sets is five.
There appears to be no clear consensus among researchers about the best
representative number of reduced sets for the amino acids, although according to our
literature survey it appears to be five. Some put it at five (Dayhoff et al., 1978, Wolynes,
1997, Wang and Wang, 1999, Cieplak et al., 2001, Koisol et al., 2004, Rakshit and
Ananthasuresh, 2008), some at six (Ptitsyn and Ting, 1999, Mirny and Shakhnovich,
1999, Mirny and Shakhnovich, 2001), seven (Plaxco et al., 1995, Bradley et al., 2002)
and even ten (Murphy et al., 2000, Fan and Wang, 2003, Li et al., 2003).
2.2 Computational de novo protein sequence design
The sequence design problem may be stated as: given a protein conformation, find the
best set of sequences that will preferentially fold to that conformation. Thus, if a protein
conformation consists of N residues, there will be 20N possible sequences for the
stipulated conformation. The exhaustive enumeration and evaluation of the sequence
space of a protein is still beyond the reach of modern computing power (Floudas et al.,
2006). However, there is an implicit consensus among researchers in this field that the
actual set of sequences that will fold to a given protein structure and be stable in that
structure, i.e., not fold to any other structure, is a very small set of the sequence space of
that protein (Saven, 2002, Xia and Levitt, 2004). This has been the guiding motive behind
the development of most computational methods for protein sequence design.
The sequence design problem, also known as “the inverse folding problem”, and
its complexity were first outlined by Drexler (1981). Ponder and Richards (1987)
developed an algorithm that could select sequences preferentially for a protein structure
core based on fixed tertiary templates. They first developed rotamer library for protein
sequences, which was later incorporated by some research groups (Hellinga and
Richards, 1994, Kono and Doi, 1994, Desjarlais and handel, 1995, Harbury et al., 1995,
Dahiyat and Mayo, 1996, 1997, DeMaeyer et al., 1997, Lazar et al., 1997, Malakauskas
and Mayo, 1998, Koehl and Levitt 1999,a,b, Raha et al., 2000, Moffet and Hecht, 2001,

23
Larson et al., 2002) as an essential tool for de novo protein design. Bowie et al. (1991)
developed a novel scoring function for amino acid residues for designing sequences of
known protein backbones. Their scoring function was based on the environment of the
residues in each protein structure.
Yue and Dill (1992) raised the question of finding good sequences that fold to a
target structure as native conformation. In their work, they asserted on the issue of
stability of the designed sequence, i.e., sequences that will fold to the target structure as
native conformation of lowest accessible free energy and simultaneously not fold into
other structures of the same or lower free energy. They developed a heuristic technique
for hydrophobic and polar residues and applied it on two-dimensional lattice models.
Their work brought forward an important conclusion, namely, a bound on the
composition of residues is essential for stability. Koehl and Levitt (1999, 2002) showed
that specificity of a designed sequence, i.e., incompatibility with competing folds is
achieved when amino acid composition is held fixed based on the approximations of the
Random Energy Model (REM) (Derrida, 1980, Shakhnovich and Gutin, 1993, Pande et
al., 1997). The general design principle for specificity is that the designed sequence
should be such that the energy gap between the target structure and other possible native
structures should be maximum. The requirement for maximization of energy gap is
formulated in terms of maximization of the Z-score (Shakhnovich and Gutin, 1993,
Abkevich et al., 1996, Mirny and Shakhnovich, 1996, Liwo et al., 1997b, Hao and
Scheraga, 1999, Lee et al., 2001).
Computational sequence design has been generally posed as a discrete search
problem because designing a sequence involves determining site-specific amino acid
residues which are discrete entities. A variety of discrete search techniques are used, the
most widely used deterministic technique being the dead-end elimination (DEE) method
(Desmet et al., 1992, Dahiyat and Mayo, 1996, 1997, Lasters et al., 1995, DeMaeyer et
al., 1997, Gordon and Mayo, 1998, Looger and Hellinga, 2001). The DEE method
systematically eliminates rotamer conformations incompatible with global energy
minimum using the dead-end elimination theorem (Desmet et al., 1992, Goldstein, 1994).
Incorporating backbone flexibility is the main drawback of DEE, as it leads to an
exponential increase in the number of rotamer conformations (Voigt et al., 2000). To

24
overcome this, some modifications have been proposed for using DEE efficiently for
protein design (Keller et al., 1995, Harbury et al. 1998, Gordon and Mayo, 1999, Pierce
et al., 2000, Wernisch et al., 2000). Another deterministic approach is based on the mean-
field theory, which incorporates the knowledge of a given set of backbone conformations
to design a potential which specifically selects sequences suitable to that set of backbone
conformations (Lee, 1994, Koehl and Delarue, 1994, Saven and Wolynes, 1997). Instead
of specifying particular sequences, this approach specifies the probabilities of different
amino acid residues at a particular position in the backbone (Saven and Wolynes, 1997,
Zou and Saven, 2000, Kono and Saven, 2001). However, since this approach is
knowledge-based, and it may contain potentials which may not have physically realistic
basis, it may face difficulties in designing sequences for de novo conformations (Thomas
and Dill, 1996, Moult, 1997, Zhang and Skolnick, 1998).
The other set of widely used techniques for de novo protein design is to search the
sequence space by sampling in a semi-random manner, which depends on algorithm
specific rules (Desjarlais and Clarke, 1998). This set consists of methods such as the
Monte Carlo Metropolis algorithm (MC) (Metropolis et al., 1953) and genetic algorithm
(GA) (Holland, 1993) and related methods. The advantages of both MC (Lee and Levitt,
1991, Hellinga and Richards, 1994, Dahiyat et al., 1997, Irbäck et al., 1998, Kuhlman et
al., 2003) and GA (Tuffery et al., 1991, Desjarlias and Handel 1995, Pedersen and Moult,
1996, Raha et al., 2000) are that both are easy to implement, to incorporate backbone
flexibility, and to design long chains. Furthermore, they do not depend on pair-wise
contribution of potential energy terms, which some (e.g., Gordon and Mayo, 1999)
believe may lead to erroneous calculations. The disadvantage of such stochastic methods
is that they may not converge to global minimum energy (Desjarlais and Clarke 1998,
Voigt et al., 2000). Hybrid methods have been developed to incorporate backbone
flexibility and to determine global minimum energy rotamer conformations without
computational deadlock. Such methods use both deterministic and stochastic search
techniques (Fung et al., 2008).
Recently, we note an interest in approaching the de novo protein sequence design
problem using gradient-based continuous optimization techniques (Koh et al., 2005 a, b,
Ananthasuresh, 2006, Jha et al., 2006, Koh et al., 2009, Jha et al., 2009). Continuous

25
gradient-based optimization is efficient in finding local minima deterministically
(Papalambros and Wilde, 2000). Using multiple initial inputs, continuous optimization
techniques can be used to efficiently search a multiple-minima problem such as the
inverse folding problem. Koh et. al. (2005) first proposed the protein sequence design
problem as a quadratic programming problem and attempted to solve it using gradient
based continuous optimization. They used the hydrophobic-hydrophilic (H-P) model for
amino acids and lattice models for protein structures. Ananthasuresh (2006) presented
different ways of posing the discrete sequence space as continuous functions which can
be solved by continuous optimization techniques. In this work, he drew analogy between
de novo sequence design and structural topology optimization problems with material
constraints. Jha et al. (2009) expanded the H-P model to a reduced five letter amino acid
alphabet and used real protein structures from Protein Data Bank (PDB). They used three
inter-residue coarse grained energy matrices to design several million minimum energy
sequences for a few proteins. Recently, Koh et al. (2009) also used the artificial power
law of gradient based topology optimization techniques to design protein sequences for a
few real proteins. The work presented in this thesis proposes two different continuous
function formulations for de novo sequence design and demonstrates their efficacy using
gradient based continuous optimization with suitable examples.
2.3 Elastic Network
The elastic network approach forms an important class of methods for analyzing the
motion of macromolecules. The normal modes of the elastic network of a protein provide
valuable information about its conformational space (Bahar and Rader, 2005).
The early works on normal mode analysis of proteins date back to 1980s (Go et
al., 1983, Brooks and Karplus, 1983, Levitt et al., 1985). In these works, the normal
modes were derived from the eigenanalysis of the Hessian matrix of the potential energy
as a function of the atomic coordinates of the proteins solved from the crystals. In these
early works, researchers realized that normal modes of the native state presented a novel
way of exploring the conformation space and dynamics of the proteins. However, the
calculation of the Hessian from the potential energy function was a computationally
daunting task and stood as a bottleneck in the normal mode analysis of large protein
structures (Tirion, 1996).

26
Tirion (1996) first proposed a single parameter harmonic potential for deriving
the normal modes of proteins and thus paved the way for computational efficiency of this
problem. His calculations showed good correlation with normal modes derived form
potential energy functions and B-factors obtained from X-ray crystal data. Bahar et al.
(1997) incorporated the random network theory of elastomers (Flory, 1976) and proposed
a Gaussian network model (GNM) for proteins. In this work (Bahar et a., 1997, Halilgolu
et al., 1997), they did eigenanalysis of the Kirchoff or the valency-adjacency matrix
(Eichinger, 1972) and showed that the normal modes of the Kirchoff matrix could be
successfully used to derive the temperature factors (B-factors) measured from X-ray
crystallographic data of protein crystals. These successes initiated the trend of normal
mode analysis using simple connectivity based matrices, thus making calculations of
normal modes for large protein structures more approachable as well as nullifying the
incorporation of experimental errors in theoretical models. The normal modes of the
Gaussian network give space-averaged fluctuation dynamics of the protein structures. To
account for anisotropy in directional fluctuations, Atilgan et al. (2001) proposed the
anisotropic network model (ANM) for proteins. Hinsen and co-workers (Hinsen, 1998,
Hinsen et al., 1999) presented a distance-dependant single parameter based elastic
network model for proteins. Coarse-grained elastic network models, which group
residues as rigid bodies (Tama et al., 2000, Li and Cui, 2002, Schuyler and Chirikjian,
2003, Bahar and Rader, 2005) or unified sites (Doruker et al., 2001, Kurkcuoglu et al.,
2004), have been proposed to analyze the motion of large proteins and supramolecular
complexes. Most of the present applications of elastic network theory on proteins are
based on GNM or ANM. Next, we present a brief overview of the wide application of
elastic network theory on the structural and functional aspect of proteins.
The success of the elastic network theory lies in capturing the functional and
domain motions of proteins using the eigenmodes of the elastic network matrix at low
computational cost compared to other methods such as molecular dynamics. The large-
scale dynamics of large supramolecular complexes like the ribosome (Tama et al., 2003,
Wang et al., 2004), GroEL (Ma and Karplus, 1998, Ma et al., 2000) and viral capsids
(Kim et al., 2003, Tama and Brooks, 2005, Rader et al., 2005), which are computationally
expensive for molecular dynamics, have been successfully simulated by elastic network

27
models. On this note, it will be pertinent to mention that the low frequency modes
derived from coarse-grained elastic network models have been used to steer molecular
dynamics simulations (Zhang et al., 2003, He et al., 2003, Tatsumi et al., 2004). Different
allosteric transitions, for example, the hinge bending motion of the transfer RNAs both in
free and bound form (Bahar and Jernigan, 1998), open/closed conformational transitions
in DNA dependant polymerases (Delarue and Sanejouand, 2002), transition of
haemoglobin from terse (T) state to relaxed (R) state (Xu et al., 2003), the hinge bending
motion of lysozyme (Brooks and Karplus, 1985, Levitt et al., 1985), etc., have been
explained using the low frequency modes of the elastic network of the corresponding
proteins. Elastic network models are also used in identifying residues that are important
for stability or are critical for folding (Micheletti et al., 2002, Rader and Bahar, 2004,
Rader et al., 2004), catalytic residues (Yang and Bahar, 2005), binding sites for receptor-
ligand complexes (Halilgolu et al., 2004, Erman, 2006, Halilgolu et al., 2008), and
deformable residues (Kovacs et al., 2004). Recently, the normal modes of elastic network
have been used to construct atomistic models of proteins from low resolution
experimental data, for example, cryo-electron microscopy (Tama et al., 2002, Delarue
and Dumas, 2004). Elastic network models have also been used in interpreting the gating
behavior of membrane proteins, for example, Gramicidin-A (Roux and Karplus, 1988),
Rhodopsin (Rader et al., 2004), potassium ion channels (Shen et al., 2002, Srivastava and
Bahar, 2006), mechanosensitive channels (Valadie et al., 2003), Nicotinic Acetylcholine
Receptor (Szarecka et al., 2007), etc.
Although, most of the applications of normal mode analysis using elastic
networks have been in analyzing functional motions around native state structures of
proteins, there are a few interesting applications of the elastic network theory in exploring
the global conformation space of proteins. Erman and Dill (2000) proposed a simplified
model of protein folding based on the equations of motion of the polypeptide. They used
Go models for polypeptides and their coarse-grained energy potential consisted of only
two components: a pairwise interaction term between residues, and an excluded volume
term acting on each residue to prevent collapse. They showed that the energy landscape
has multiple minima and the number of minima is a function of the number of
eigenvalues of their elastic network model for the polypeptides. Ball et al. (2002) further

28
proposed protein folding as a variant of the traveling salesman problem (TSP) using the
elastic network optimization strategy. Kim et al. (2003) used elastic networks to generate
transition models of protein structures between two conformational states. Miyashita et
al. (2005) used an elastic network model to explore the energy landscape between two
stable equilibrium structures of proteins. Güner et al. (2006) proposed a model for
generating optimal folding pathways of proteins based on elastic networks and optimal
control theory. Rakshit and Ananthasuresh (2010) used the normal modes of elastic
network to predict tertiary structures from unfolded states of proteins using gradient
based optimization techniques. We will discuss more about this work in section 5.3.
Other interesting application of elastic networks for proteins lie in automatic domain
decomposition (Kundu et al., 2004) and analysis of domain swapping in proteins (Kundu
and Jernigan, 2004), exploration of functional and evolutionary relations in protein
superfamilies (Leo-Macais et al., 2005) and the use of normal modes as a classifying
statistic for proteins (Krebs et al., 2002).
2.4 Minimalist coarse-grained models
Coarse-grained models and simulations have re-surfaced as important computational
tools with current emphasis on biomolecular-system simulations that span in orders of
magnitude both in the scales of space and time (Tozzini 2005, 2010). Minimalist coarse-
grained models for proteins are a sub-class of coarse-grained models which use the
“maximum level of coarsening that still allows us to explicitly represent some
fundamental feature of the bio-molecule, such as the secondary structure level” (Tozzini,
2010). The work presented in this thesis uses the simplest of the minimalist coarse-
grained models, namely the one-bead coarse-grained (OB-CG) model. In this section, we
review the literature related to OB-CG models.
One-bead models were first introduced by Gō (Ueda et al., 1978) for simplified
representation of protein structures on two and three-dimensional lattices. Since then,
lattice models have been successfully used to explore the physical-chemical properties of
proteins, for example, folding and hydrophobic collapse (Ueda et al., 1978, Abe and Gō,
1981, Dill, 1984, Yue et al., 1995, Mirny and Shakhnovich, 2001), energy funnels
(Bryngelson et al., 1995, Onuchic et al., 1997), designing and testing energy functions
(Mirny and Shakhnovich, 1996, Thomas and Dill, 1996), designing sequences (Yue and

29
Dill, 1992, Shakhnovich and Gutin, 1993), etc. The usefulness of lattice models lies in
reducing the infinite conformation space to a finite space of two or three dimensions.
Thus, using lattice models, the conformation space of a protein can be extensively
searched. However, although lattice models have been used to infer important properties
of proteins and can map real three-dimensional structure of small proteins onto lattice
structures, they cannot do so for medium or large proteins that demonstrate hierarchical
levels in structure.
Off-lattice one-bead models are the simplest of the minimalist coarse-grained
models based on realistic protein structures. Each bead in the OB-CG model represents a
residue in the polypeptide chain. The OB-CG models can be of different types depending
on the position of the representative bead relative to the backbone of the protein. The
most widely used schemes are the ones that place the bead on the C-α coordinates of the
backbone. This model has several advantages with respect to interchangeability with
experimental data (Trylska et al., 2005) and simplicity in representation of the force field
terms (Tozzini, 2010). However, representations of force field terms which depend on the
volume of each amino acid (e.g., the excluded volume effect) become complicated with
such OB-CG models. Other OB-CG models place the interacting bead on the C- β
coordinates or the centroid of each amino acid residue. Depending on the nature of
simulation, researchers have used models which use reductionist approach for higher
levels in proteins, for example, secondary structures (Erman et al., 1997, Nanias et al.,
2003, Sancho et al., 2004, Yue and Dill, 2008), tertiary folds (Doruker et al., 2001,
Schuyler and Chirikjian, 2003, Bahar and Rader, 2005) and even whole proteins (Tozzini,
2010).
Proteins fold under the action of complex interplay of a number of molecular
forces, the dominant ones being entropic (hydrophobic-hydrophilic interactions with
solvent), hydrogen bonds, disulphide bonds, salt bridges, electrostatics, van der Waals
interactions, etc. The process of protein folding is so complex that determining the final
protein structure, i.e., the tertiary or quaternary structure, from the sequence is regarded
as the holy grail (Klepeis and Floudas, 2003) of computational biology and chemistry.
However, considering the protein structures that exist in nature (referred to as the native-
state) are the most optimal ones with respect to folding and hence energy (Anfinsen,

30
1961, 1973), inter-residue coarse-grained potentials that consider amino acid residues as
single-point interacting entities i.e., one-bead can be developed (Tanaka and Scheraga,
1976). The most well-known among such coarse-grained structure-derived potentials is
the Miyazawa-Jernigan (MJ) matrix (Miyazawa and Jernigan, 1985). In their classic
work, Miyazawa and Jernigan used the Boltzman inversion technique to derive potentials
from the statistics of inter-residue contacts from protein crystal structures. They
incorporated solvent interaction of the amino acid residues by introducing a random
mixing model based on the quasi-chemical approximation (Hill, 1986) for calculating the
reference state. Their original work (1985) was based only on 42 globular protein
structures; subsequently they re-evaluated their inter-residue potential matrix based on
1661 globular protein structures (Miyazawa and Jernigan, 1996). It is interesting to note
that there is little difference between the inter-residue potential matrix derived in 1985
and that derived in 1996, which underscores the robustness of their calculations. Other
notable coarse-grained structure-derived potentials include the distance-dependant
potential proposed by Sippl (1990) and potentials of mean-force derived for maximizing
Z-scores for a set of native structures of nonhomologous proteins (Mirny and
Shakhnovich, 1996).
Apart from structurally derived potentials, considerable research effort has gone
into deriving potentials on more physically realistic energies, for example, electrostatics,
van der Waals, hydrogen bonds, disulphide bonds, explicit interaction with solvent
molecules, etc., and which form the basis for more abinitio simulations, for example,
molecular dynamics. Since the work in this thesis is concerned with OB-CG models, we
will only discuss such coarse-grained models, although extensive literature exists on
multi-level coarse-grained models and atomistic potentials (Tozzini, 2010). The first of
such force fields was proposed by Levitt and Warshel (1975). In this work, they
introduced a coarse-grained model of proteins in which each residue in the backbone was
represented by the corresponding C-α and the centroid coordinates of the side chain and
the only degree of freedom corresponding to each residue was the torsion angle about the
line joining two adjacent C-α coordinates. Their folding model was based on space-
averaged forces derived from a Leonard-Jonnes type potential and interactions of side
chains with solvent. Levitt (1976) extended this work to include more energy terms,

31
namely, disulphide and hydrogen bond terms and interactions with near neighbors. The
hydrogen bond term was calculated by introducing pseudo coordinates for backbone
Oxygen and Nitrogen atoms based on C-α coordinates without introducing any
additional variables. However, this coarse-grained energy model contains two terms
which have much less physically realistic foundation than other energy terms, namely,
the ‘holding’ and ‘pushing’ potentials, which were most probably introduced to enhance
numerical accuracy and overcome issues related to the particular numerical scheme used
in that work (Levitt, 1976). Another notable OB-CG model developed on the basis of
physical forces is the UNRES force field developed by Scheraga and co-workers (Liwo et
al., 1997 a, b).
In the work presented in this thesis, we explore the tertiary conformation space of
proteins using OB-CG models. We incorporate the inter-residue contact energies given
by the MJ matrix (Miyazawa and Jernigan, 1996) into a continuous function (Rakshit and
Ananthasuresh, 2010) and use it to predict tertiary conformations. We also adopt a few
terms of Levitt’s coarse-grained potential (Levitt, 1976) for tertiary structure predictions.
2.5 Closure
In this chapter we presented literature review on the related computational techniques
which we developed for our goal of simultaneous search in sequence and conformation
spaces. In the first section, we present the relevant works on reducing the amino acid set,
the different computational techniques on which these works depend, and the
computational technique that we adopt. The second section describes different
computational methods for protein sequence design, and highlights a few earlier works
on sequence design using continuous optimization approaches which we developed
further in the work presented in this thesis. In the third section we present literature
review on elastic networks and its diverse applications. The fourth section contains
literature review on coarse-grained models used in protein folding simulations relevant to
the work presented in this thesis.

32
3. Reduced Amino Acid Alphabet using Metric
Multi-dimensional Scaling (MMDS)
• We present the motivation behind reducing the amino acid set and using MMDS.
• The method of MMDS and the derivation of a low dimensional map from a set of
interconnected data are described.
• We use MMDS on the MJ matrix and present the results with suitable discussion.
• The chapter is closed by a brief summary.
3.1 Introduction
In this work we present a map based on the inter-residue contact energies given by the
Miyazawa-Jernigan (MJ) matrix (Miyazawa and Jernigan, 1996) using metric multi-
dimensional scaling (MMDS) (Kruskal, 1978). By presenting the data in a visual form,
we hope to reduce the complexity of finding out the inter-relations among the residues
which might not be directly evident from the MJ matrix. Each amino acid is represented
as a point on the MMDS map. The distance between two points on the map quantifies the
dissimilarity in their contact energies. The larger the distance the larger the dissimilarity.
This map elucidates relationships among the amino acids that are not easily discerned
from the MJ matrix.
The MMDS method is frequently used for a visual representation from a set of
data representing the relation among a number of objects. Similar work was reported by
French and Robson (1983) who had derived a map using MMDS for amino acids from
Dayhoff’s “relatedness odds matrix” (1972). The MMDS map presented in this chapter
verifies that hydrophobicity is the key feature that characterizes the amino acid residues
and the inter-residue contact energies represent a rough hydrophobicity scale (Cornette et
al., 1987, Chan, 1999, Venkatarajan and Braunn, 2001). Additionally, with the help of
this map, we compare (the similarities/differences among amino acid residues as
represented by) the MJ matrix with Block Substitution Matrix (BLOSUM) 62 (Heinkoff
and Heinkoff, 1992) and Pointwise Accepted Mutations (PAM) (Schwartz and Dayhoff,
1978) 250 matrices.

33
A novel feature of our map is that it can be used as a visual method of reducing
the amino acid set. We support this by determining the groups using a hierarchical
clustering method (Johnson and Wichern, 2006). We are also able to arrive at an
optimum number of groups for reducing the amino acid set by using this method.
3.2 Method
Metric Multi Dimensional Scaling (Mead, 1992) is a multi-variate statistical analysis
technique that is used for making a visual representation from a n n× matrix representing
the interaction between a set of n objects that one is interested to study. The thij entry in
the matrix represents the interaction between thi and thj objects. If the thij entry in the
matrix represents dissimilarity between the thi and thj objects, then the matrix is called
the dissimilarity or distance or proximity matrix. Here, as there can be no dissimilarity
between an object with itself all the diagonal elements are zero. On the other hand, if the
thij entry into the matrix represents similarity between the thi and thj objects, then the
matrix is called the similarity matrix. In this case, the diagonal elements are non-zero.
The results are represented as a plot of n points representing the n objects on a space of
two or higher dimensions. This method was first suggested by Torgerson (1952) and then
developed and used by Kruskal and Wish (1964) in representing as varied and qualitative
things as cultural similarity among nations and dialect of Salish Indians. More recently,
this map was used for classifying engineering materials based on ergonomic and aesthetic
considerations (Ashby and Johnson, 2002).
The key feature of MMDS method is that it reveals the hidden structure among
the objects that lies buried in the mass of data stored in a matrix form. Similar points are
huddled together in the plot and the distances among the points give a measure of
similarity among the objects. Furthermore, one can often identify variation of key
parameters on which these objects depend along different directions in the map.
Mathematically, constructing an MMDS map can be shown to be a least-square
minimization problem. Let n objects be represented by a set of n points on a plane. Let
the distance between thi and thj points be ij
d and its corresponding entry in the

34
proximity matrix is ij
δ . The MMDS technique attempts to minimize all such distances in
the sense of least squares, i.e. ,
( )( )2
2
,, 1
n
ij ij
i ji j
Minimize d δ=
≠
−∑x y
(3.1)
where,
1 2{ , , , }nx x x=x � and 1 2{ , , , }ny y y=y � are the x and y coordinates of the n points in
the map, and
( ) ( ) ( )2 22
ij i j i jd x x y y= − + − (3.2)
where the superscript in braces indicates the dimensionality of the MMDS map (in this
case it is two as we have chosen a planar representation). Therefore, we can write Eq. 3.1
as,
( ) ( )2
2 2
,, 1
n
i j i j ij
i ji j
Minimize x x y y δ=
≠
− + − −
∑
x y (3.3)
The solution of the minimization problem in Eq. 3.3 gives the coordinates of the points
and helps create the MMDS map. It should be noted that the MMDS map is unaffected
by the orientation of the chosen coordinate system, i.e., the final set of points may be
oriented differently in different runs with different initial guesses but the relative
positions of the points do not change. This happens because MMDS deals with only the
distances between the points which are devoid of any directional information. We have
used MATLAB’s optimization toolbox program fminunc (unconstrained optimization
which uses sequential quadratic programming combined with trust region method) to
solve the above least-square minimization problem in constructing the map. However, the
thij entry of the MJ matrix cannot be directly used as ijδ in Eq. 3.3. The treatment of the
MJ matrix to get the ijδ s is discussed next.
The thij entry in the MJ matrix represents the contact energy between thi and thj
amino acids. The diagonal entries represent contact energy between same amino acids.
Therefore, the extent to which the thij entry matches the corresponding diagonal entries
(both thi and thj diagonal entries) represents the similarity between the th
i and thj amino

35
acids. Thus, the MJ matrix can be taken as a similarity matrix. To convert the MJ matrix
to a proximity matrix we do the following operation:
2
ii jj
ij ij
M MMδ
+ = −
(3.4)
where , , and ii ii ijM M M are the , , and th th thii jj ij entries in the MJ matrix respectively.
Here, we take the absolute value as ijδ represents distance between two points and hence
is always positive. This symmetric transformation ensures that all the diagonal entries are
zero. With this matrix now one can select multiple dimensions (one and above) for
minimizing Eq. 3.3. For a given dimension it may not be possible to position all the
points on the map such that the distances among them exactly match the corresponding
distances given by the proximity matrix. The extent to which it deviates from the actual
data is given by a measure called stress (Kruskal and Wish, 1964). This stress is given
by,
( )( )2
2( )
q
ij ij
i j
i j
ij
i j
i j
d
Stress q
δ
δ<
<
−
=
∑∑
∑∑ (3.5)
where, q is the dimension of MMDS map. For example, when q is two ijd is given by
Eq. 3.2. The values of stress calculated for one, two and three dimensions are shown in
Fig. 3.1. We selected two as the dimension because stress is lowest there. We did not
Fig. 3.1. Plot of stress against number of dimensions
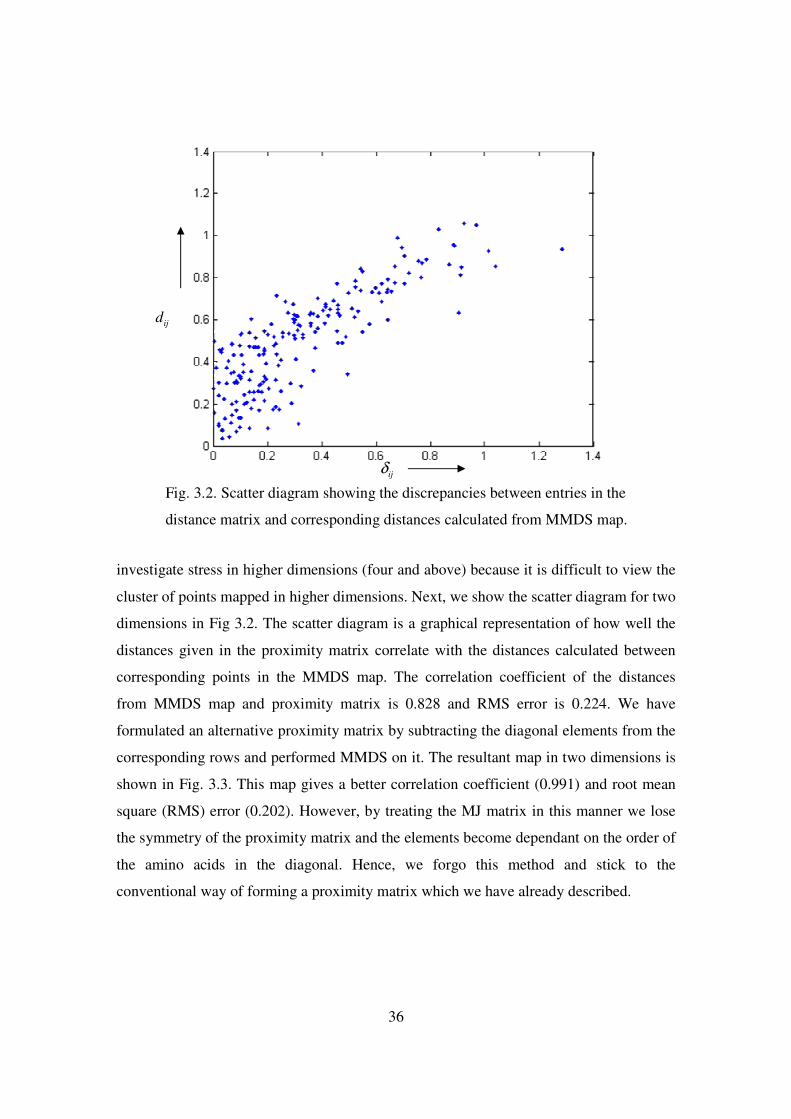
36
investigate stress in higher dimensions (four and above) because it is difficult to view the
cluster of points mapped in higher dimensions. Next, we show the scatter diagram for two
dimensions in Fig 3.2. The scatter diagram is a graphical representation of how well the
distances given in the proximity matrix correlate with the distances calculated between
corresponding points in the MMDS map. The correlation coefficient of the distances
from MMDS map and proximity matrix is 0.828 and RMS error is 0.224. We have
formulated an alternative proximity matrix by subtracting the diagonal elements from the
corresponding rows and performed MMDS on it. The resultant map in two dimensions is
shown in Fig. 3.3. This map gives a better correlation coefficient (0.991) and root mean
square (RMS) error (0.202). However, by treating the MJ matrix in this manner we lose
the symmetry of the proximity matrix and the elements become dependant on the order of
the amino acids in the diagonal. Hence, we forgo this method and stick to the
conventional way of forming a proximity matrix which we have already described.
ijd
ijδ
Fig. 3.2. Scatter diagram showing the discrepancies between entries in the
distance matrix and corresponding distances calculated from MMDS map.
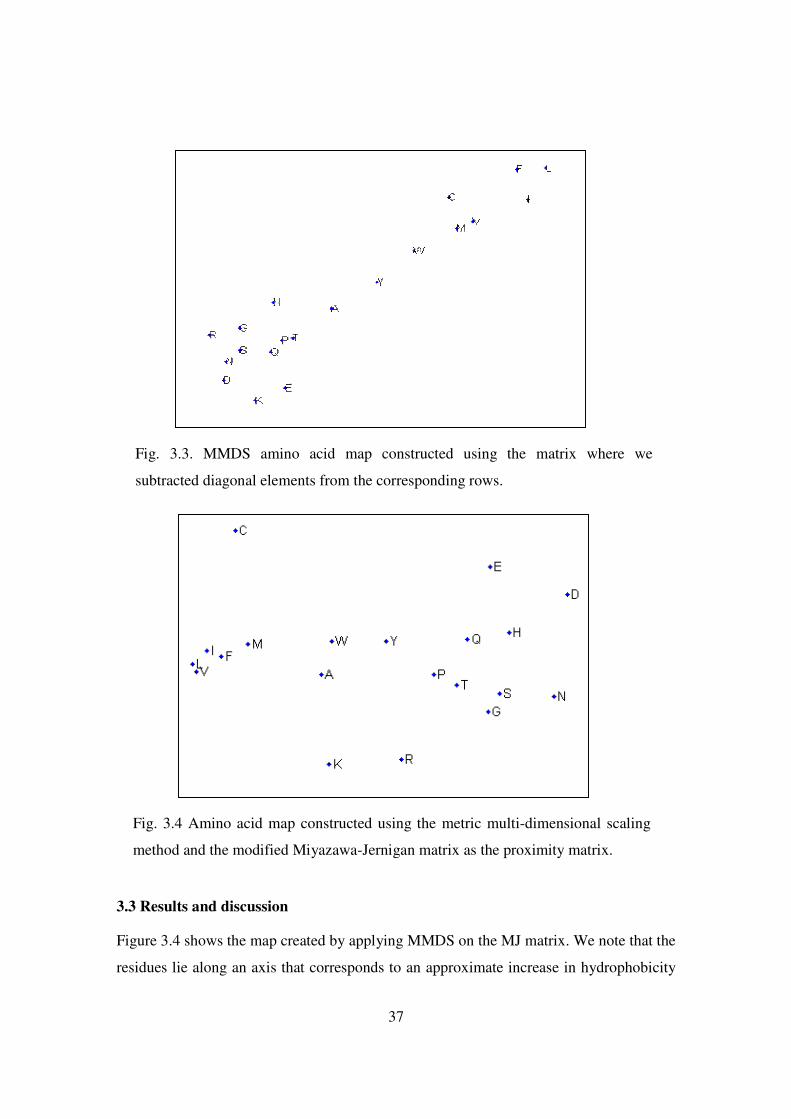
37
3.3 Results and discussion
Figure 3.4 shows the map created by applying MMDS on the MJ matrix. We note that the
residues lie along an axis that corresponds to an approximate increase in hydrophobicity
Fig. 3.4 Amino acid map constructed using the metric multi-dimensional scaling
method and the modified Miyazawa-Jernigan matrix as the proximity matrix.
Fig. 3.3. MMDS amino acid map constructed using the matrix where we
subtracted diagonal elements from the corresponding rows.

38
(Cornette et al., 1987). This axis is shown in Fig. 3.5. The curved axis in Fig. 3.5 shows
the direction of increase in inter-residue contact energies. We also show the classification
of amino acids according to their chemical properties as done by Dayhoff (1972) in this
figure.
In Fig. 3.6, we show the residues that favorably substitute one another in the
BLOSUM database on the map. All the residues that have a positive log odd score in the
BLOSUM62 matrix are connected by double ended arrows in this figure. This figure
shows that both substitutionally (BLOSUM62) and energetically (MJ matrix), Cystine
stands separate from other amino acid residues. The hydrophobic residues and the
hydrophilic ones do not substitute one another favorably. This substitution is also
unfavorable from contact energy viewpoint as shown by the map. According to the map
Proline, Threonine and Glutamic acid, being near to one another, should be favorable for
substitution; this inference is not supported by BLOSUM. However, our conclusion can
be supported from the viewpoint of conservation of molecular volume in evolutionary
substitution (French and Robson, 1983). Proline, Threonine and Glutamic acid can be
grouped together in one class characterized by their smallness of volume (Schluz and
Schirmer,1978). Figure 3.7 shows the residues that substitute one another favorably in the
Percent Accepted Mutations (PAM) matrix. We connect the residues that have a positive
log odd score in the PAM250 matrix by double ended arrows. Here too, we see that
Cystine stands separate form all other amino acids in terms of evolutionary substitution
(PAM250) and inter-residue contact energy (MJ). The hydrophobic and hydrophilic
residues are likely to have different lineages in evolution as they do not substitute one
another favorably (Dayhoff et al. 1972, Miyata et al. 1979). Here, we feel that it is worth
mentioning that this map represents a unique and a novel way of drawing conclusions
from two different data sets related to the amino acid residues, namely the block
substitution (BLOSUM) or evolutionary (PAM 250) data and inter-residues contact
energy data derived from the experimental data (MJ).
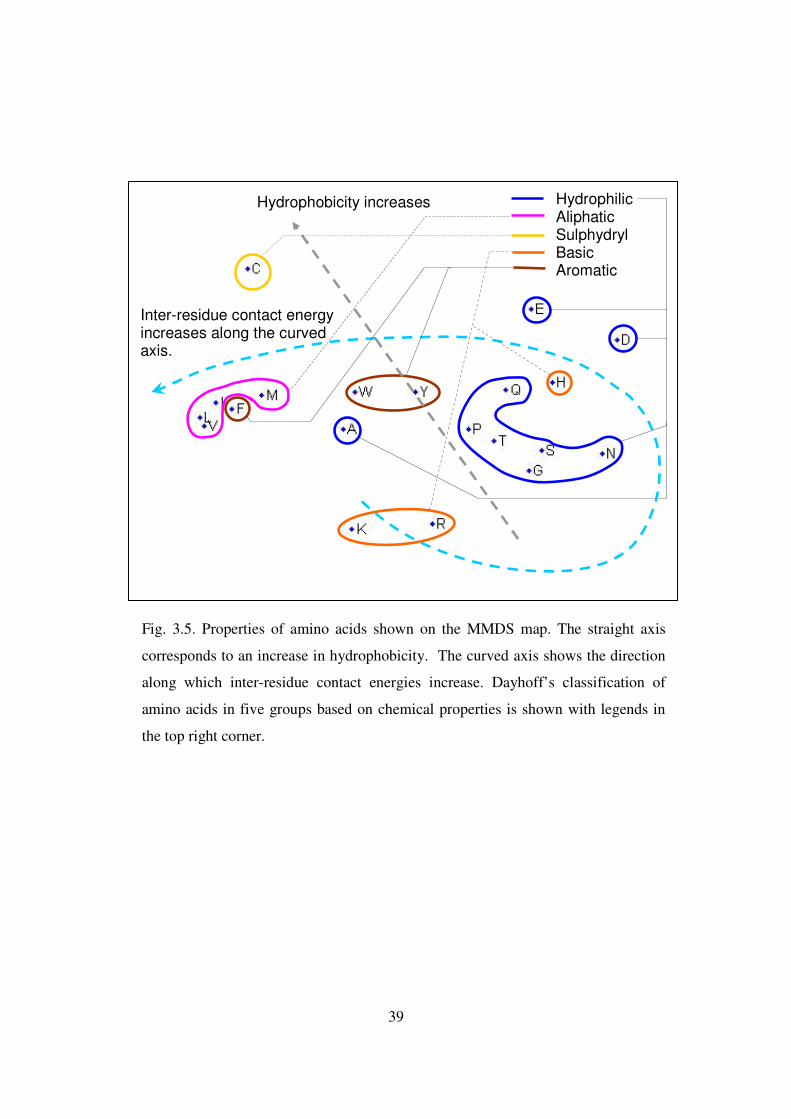
39
Fig. 3.5. Properties of amino acids shown on the MMDS map. The straight axis
corresponds to an increase in hydrophobicity. The curved axis shows the direction
along which inter-residue contact energies increase. Dayhoff’s classification of
amino acids in five groups based on chemical properties is shown with legends in
the top right corner.
Inter-residue contact energy increases along the curved axis.
Hydrophobicity increases Hydrophilic Aliphatic Sulphydryl Basic Aromatic

40
Fig. 3.7. The residues that have a positive log odd score in the PAM250
matrix are connected by double ended arrows.
Fig. 3.6. The residues that have a positive log odd score in the BLOSUM62
matrix are connected by double ended arrows.

41
Reducing the amino acid residues to a small set is a topic of active interest among protein
researchers. A few works based on the MJ matrix exist in the current literature (Wang
and Wang, 2002 and 1999, Cieplak et al., 2001, Li et al., 1997). All these works employ
different methods for reducing the amino acid set. The MMDS method provides an easy
visual method of grouping (see Fig. 3.4). To reinforce this, we use a hierarchical
clustering method based on average distance of clusters (Johnson and Wichern, 2006) on
our distance matrix to simplify the amino acid set. In this method, we find the minimum
distance in the distance matrix and group the corresponding amino acids. Next, we find
the distance between this group and all other amino acids by calculating the mean
distance from this group to all other amino acids or groups. Thus, if L (Leucine) and I
(Isoleucine) are clubbed together to form a group {L,I}, then the distance { },L I Gd of this
group from G (Glycine) is given by 2
LG IGd d+
. We continue this procedure starting
from 20 amino acids and go on grouping until we arrive at a single group. The resulting
dendrogram is shown in Fig. 3.8.
In Fig. 3.9 we plot the minimum distance between the groups as we go on
decreasing the number of groups. We see that the highest ratio of increase in the
minimum distance to the current minimum distance occurs when the number of clusters
changes from 19 to 18 and from five to four or four to three. A sudden increase in
minimum distance indicates that the groups are losing their compact size as they are
merged together. Since 18 is a large number for reducing the amino acid set we conclude
that five or four is the best number for simplifying the amino acid set. Our conclusion is
further supported by the fact that grouping amino acid residues into five sets is most
common in the literature (Dayhoff et al., 1972, Li et al., 1997, Wolynes, 1997, Murphy et
al., 2000, Wang and Wang, 2002 and 1999, Cieplak et al., 2001, Cannata et al., 2002, Li
et al., 2003, Koisol et al., 2004). In Fig. 3.10 we show these five groupings on our
MMDS map. Although our grouping is based on hierarchical clustering and our database
is contact energies from a statistical database we find distinctive chemical properties
within each group. D and E are acidic whereas K and R have basic properties. Q, H, P, T,

42
Fig. 3.8. Dendrogram showing hierarchical grouping of amino acids based on our distance matrix.
CMFILVWYAGTSNQDEHRKP
C MFILVWYAGTSNQDEHRKP
C MFILVWYAKR GTSNQDEHP
C MFILVWYA KR GTSNQDEHP
C MFILVWYA KR GTSNQHP DE
C KR WYA MFILV DE GTSNQHP
C KR WYA MFILV DE QH GTSNP
C KR WYA MFILV D QH GTSNP E
C KR D QH TP WYA MFILV E GSN
C KR D QH TP WY MFILV E GSN A
C K D QH TP WY MFILV E GSN A R
C K D QH TP
W
E GS
Y
R A MFILV WY N
C K D QH TP E GS R A MFILV
N
W Y C K D QH TP E GS R A FILV
N M
W Y C K D Q TP E GS R A FILV
N M H
W Y C K D Q TP E G R A FILV
N M H S
W Y C K D Q TP E G R A FI
N M H S LV
W Y C K D Q T E R A FI N
M H S LV P G
W Y C K D Q T E R A F N
M H S LV P G I
W Y C K D Q T E R A F N
M H S L P G I V

43
S, G, N all have small molecular volume and are hydrophilic in nature. On the other
hand L, V, I, M, F, W, Y (all except A in that group) are characterized by their largeness
in size and hydrophobic nature. Lastly, C stands alone because of its unique ability to
form disulphide bonds.
Fig. 3.9. Minimum distance between groups as a function of the number of groups.
The ratio of increase in minimum distance (between groups) as we reduce the
number of groups to the minimum distance (between groups) in current number of
groups is highest for 18 and five groups.
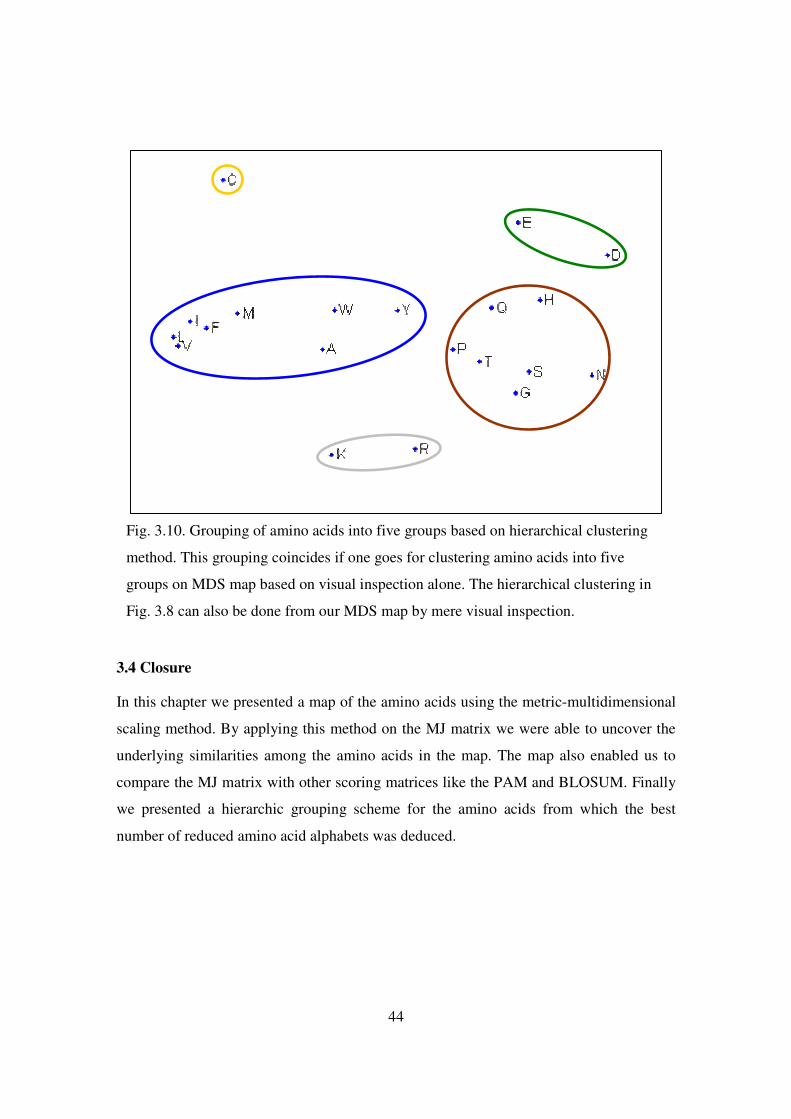
44
3.4 Closure
In this chapter we presented a map of the amino acids using the metric-multidimensional
scaling method. By applying this method on the MJ matrix we were able to uncover the
underlying similarities among the amino acids in the map. The map also enabled us to
compare the MJ matrix with other scoring matrices like the PAM and BLOSUM. Finally
we presented a hierarchic grouping scheme for the amino acids from which the best
number of reduced amino acid alphabets was deduced.
Fig. 3.10. Grouping of amino acids into five groups based on hierarchical clustering
method. This grouping coincides if one goes for clustering amino acids into five
groups on MDS map based on visual inspection alone. The hierarchical clustering in
Fig. 3.8 can also be done from our MDS map by mere visual inspection.

45
4. Search in the Sequence Space
• We give an introduction to protein sequence design for fixed backbone and its
relevance in simultaneous sequence and conformation search.
• We describe sequence design using double sigmoid interpolation technique and
apply it to design sequences for four proteins.
• We present sequence design using quadratic programming and test it on four
proteins.
• We compare the two methods and find out the relative advantages and drawbacks.
• We close the section by a brief summary.
4.1 Introduction
The sequence design problem, also known as “the inverse folding problem” (Drexler,
1981) has gained as much importance as the “protein folding problem” with recent
successes in the development of de-novo proteins (Hecht et al., 1990, Desjarlais and
Handel, 1995, Dahiyat and Mayo, 1996, 1997, Dahiyat et al., 1997, Street and Mayo,
1999, Dantas et al., 2003, Kuhlman et al., 2003, Offredi et al., 2003, Butterfoss and
Kuhlman, 2006). De-novo proteins with new folds (Farinas and Regan, 2003), higher
stability (Gillespie et al., 2003) and faster folding rates (Kuhlman et al., 2002, Dantas et
al., 2003) are giving new insights into the intricate mechanism by which proteins fold in-
vivo. A key to the success of designing a de-novo protein lies in the accurate specification
of sequences that will fold to a given target structure. With the help of computational
tools geared towards searching the vast sequence space efficiently, experimentalists can
save much effort and cost.
As discussed in the literature review section, to the best of our knowledge, all
computational sequence design techniques use discrete or heuristic optimization methods
for searching the sequence space. As mentioned earlier, our work is motivated by the
recent works of a few researchers (Koh et al. 2005, Koh et al. 2005, Ananthasuresh 2006,
Jha et al. 2006, Koh et al. 2009, Jha et al. 2009) in formulating protein sequence design
as a continuous optimization problem. In this work, we propose a novel continuous
formulation for protein sequence design using the double sigmoid function using

46
different potential functions (statistical and atomistic) and also extend the quadratic
programming formulation for protein sequence design (Koh et al., 2005) to all 20 amino
acids using atomistic potentials.
We demonstrate our methods on a set of four proteins, namely, homeodomain
chain C, PDB (Bernstein et al., 1977) ID 1HDD (Kissinger et al., 1990); calbindin, PDB
ID 4ICB (Svensson et al., 1992); protein A of human Fc fragment chain C, PDB ID 1FC2
(Deisenhofer, 1981) and Cro repressor, PDB ID 2CRO (Mondragon, et al. 1989b). The
native structures of these proteins are shown in Fig. 4.1. Our selection is based on the
FISA decoy set (Simons et al., 1997), which contains 500 decoy structures for each of the
2CRO 65 residues
1FC2 (chain C) 43 residues
1HDD (chain C) 57 residues
4ICB 76 residues
Fig. 4.1. Native structure of the four proteins that we target for sequence design.
The number of residues in each protein is also indicated.

47
aforementioned four proteins. We have used gradient-based large-scale nonlinear
optimization problem solver IPOPT (Wächter, 2002, Wächter and Biegler, 2004) (Please
refer to Appendix B1 for a short discussion on IPOPT) to design minimum energy
sequences for the abovementioned proteins. We align the designed sequences with the
wild-type sequences and find the site-specific amino acid residue matches without
inserting any gaps. The criterion for finding matches without inserting gaps is a stringent
criterion. Many sequence alignment programs align sequences by inserting gaps within
them. We, however, follow a different path by aligning sequences without gaps and
develop a novel scoring method by not only counting site-specific matches but also the
same based on simplified amino acid alphabet (Dayhoff et al., 1978, Wang and Wang,
1999, 2002, Murphy et al., 2000, Cieplak et al., 2001, Cannata et al., 2002, Li et al.,
2003, Koisol et al., 2004, Rakshit and Ananthasuresh, 2008). Reduced amino acid
alignment has been used for checking the designability of sequences (Brown et al., 2003).
Our results are satisfactory both for non-reduced and reduced amino acid set. When we
align our design sequences with the wild-type ones by using the CLUSTAL (version
1.83) (Higgins and Sharp, 1988), a software that inserts gaps for maximum alignment, we
find the number of matches increase, as expected. We also test our designed sequences by
threading them on the decoy set FISA for the aforementioned proteins (Simons et al.,
1997).
Before proceeding to the description of our sequence design methods, we would
like to specifically mention the scope of our sequence design formulations. Both the
formulations described here are based on the principle of minimization of free energy,
which governs protein folding (Anfinsen, 1961, 1973). Although wild-type sequences are
the outcomes of a complex interplay of various selective pressures, minimization of free
energy plays an important role in selecting them (Dill and Chan, 1997). Furthermore,
Shakhnovich and Gutin (1993) used lattice models to demonstrate that the minimum
energy sequences are also the ones that fold at faster rate than random sequences.
However, free energy minimization is not the best method for designing de novo protein
sequences, especially based on statistical potentials (Yue et al., 1995, Thomas and Dill,
1996, Moult, 1997, Zhang and Skolnick, 1998). Based on structural information of the
native-state, formulations have been developed to design sequences to maximize the Z-

48
score, i.e., maximization of the energy gap between native and competing structures and
minimization of the fluctuation of energy over all native-like structures (Deutsch and
Kurosky, 1996, Morrissey and Shakhnovich, 1996, Seno et al., 1996, Mirny and
Shakhnovich, 1996). Z-score optimization has also been used to calculate statistical
potentials or statistically tuned physical potentials specifically for guiding designed
sequences to fold to target structures (Koehl and Delarue, 1996, Dahiyat and Mayo, 1997,
Chiu and Goldstein, 1998, Zhang and Skolnick, 1998, Gordon and Mayo, 1999, Mendes
et al., 2002, Dokholyan, 2004, Gordon et al., 1999, Liang and Grishin, 2004, Pokala and
Handel, 2005, Alvizo and Mayo, 2008). However, in this work, we do simultaneous
search in sequence and conformation spaces. Z-score based potentials or Z-score based
formulations are not amenable to use when both sequence and conformation are
unknown. For this reason, we base our formulations on minimization of free energy
rather than Z-score.
In the first phase of simultaneous sequence and conformation search, when both
are unknown, we use statistical potentials to design sequences (our simultaneous
sequence and conformation search strategy will be described in detail in Chapter 6). The
applicability of statistical potentials has been argued for de novo sequence design (Yue et
al., 1995, Thomas and Dill 1996, Moult 1997, Zhang and Skolnick 1998). Hence, in the
subsequent sections where we evaluate our sequence design formulations, we present the
best designed sequences not on the basis of the energy of the designed sequences, but by
matching them against the wild-type sequences and also by calculating the energy gap
and dispersion of energy of the designed sequences. Thus, the above two criteria, i.e., the
number of matches with wild-type sequences and the calculation of the energy gap and
dispersion of energies based on decoy sets, serve as alternative tests for our formulation
of sequence design based on continuous optimization.
The rest of this chapter is organized as follows. We next describe our sequence
design formulations using the double sigmoid function. Thereafter, we present the results
using this method. In the following section, we describe sequence design formulation
using quadratic programming. After that, we present the results using quadratic
programming approach on the same set of proteins. We end the chapter with a discussion
of both the methods.

49
4.2 The Double Sigmoid method
In this section we describe the formulation of protein sequence design with a fixed
backbone using the double sigmoid function for interpolating the energy. To demonstrate
the generality of our approach, we use three different potentials for energy calculation.
First, we consider the Miyazawa Jernigan (MJ) potential (Miyazawa and Jernigan, 1996),
which is a coarse grained statistical potential based on inter-residue contacts at the
tertiary level. Next, we use the Zhang Skolnick (ZSk) potential (Zhang and Skolnick,
1998), which not only gives statistical potential based on inter-residue contacts at the
tertiary level, but also includes a separate scoring table for the propensity of amino acids
for secondary structures like alpha helices and beta strands. Finally, we use a full
atomistic potential which consists of atomistic van der Waals and electrostatic potentials
of the AMBER force field (Cornell at al., 1995) and the implicit mean solvation force
field calculated by Fraternali and Gunsteren (1996). Later, we also describe a formulation
for imposing composition constraint on designed protein sequences.
4.2.1 Formulation of the Continuous Optimization problem
Consider an N-residue protein whose structure is given and a sequence is to be designed
for it. If we consider ix as the design variable corresponding to the ith residue position,
we will then have { }: 0 20, 1,ix i N≤ ≤ =x as the vector of design variables (Fig. 4.2a).
The bound on ix is due to the number of amino acids; thus, 0 1ix≤ < represents Alanine,
1 2ix≤ < represents Cystine and so on as we map the amino acids on the real line �
bounded between 0 and 20. It is to be noted that the order of the amino acids on the real
line is of no consequence, but, once a certain order has been fixed, it has to be followed
for all ix s. We now give the simple formulation for the interaction between residues if
there are only two types of residues, say H (hydrophobic) and P (polar). As there are only
two types of residues in this case, { }: 0 2, 1,ix i N≤ ≤ =x . Thus, 0 1ix≤ < represents H
and 1 2ix≤ < represents P. Let the interaction energy matrix be given by,
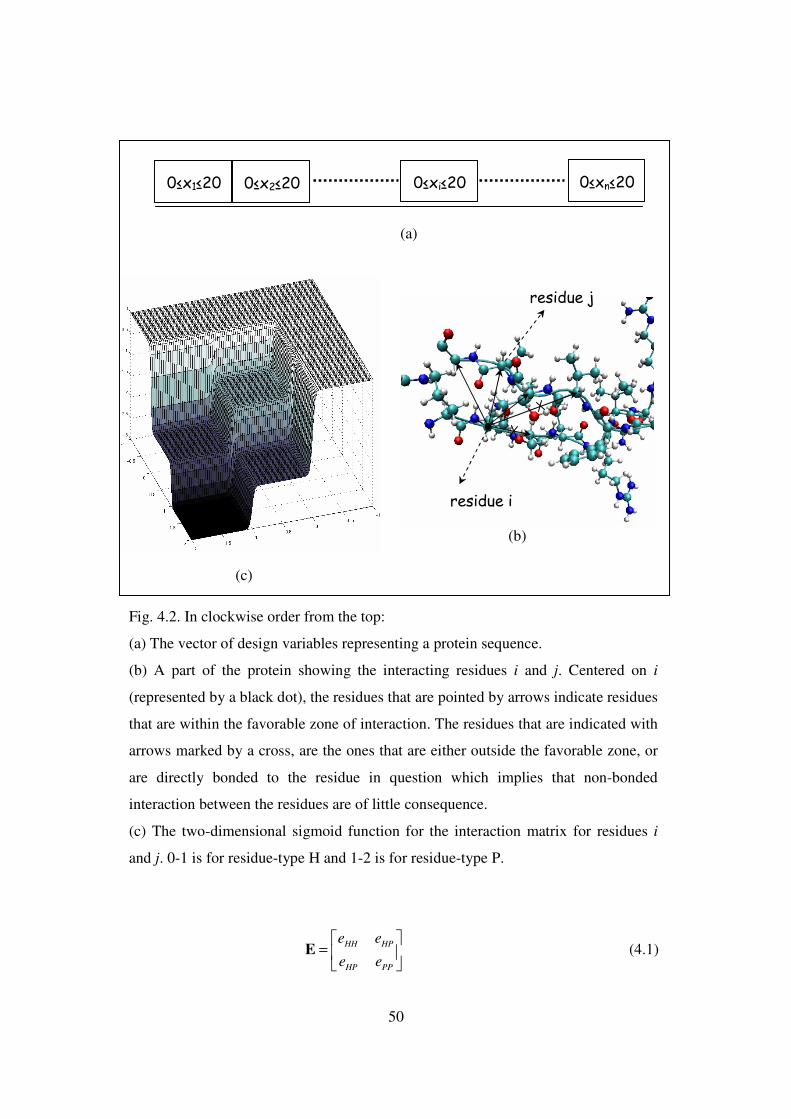
50
HH HP
HP PP
e e
e e
=
E (4.1)
Fig. 4.2. In clockwise order from the top:
(a) The vector of design variables representing a protein sequence.
(b) A part of the protein showing the interacting residues i and j. Centered on i
(represented by a black dot), the residues that are pointed by arrows indicate residues
that are within the favorable zone of interaction. The residues that are indicated with
arrows marked by a cross, are the ones that are either outside the favorable zone, or
are directly bonded to the residue in question which implies that non-bonded
interaction between the residues are of little consequence.
(c) The two-dimensional sigmoid function for the interaction matrix for residues i
and j. 0-1 is for residue-type H and 1-2 is for residue-type P.
0≤x1≤20 0≤x2≤20 0≤xi≤20 0≤xn≤20
(a)
residue i
residue j
(b)
(c)

51
When residues i and j are within the interaction distance as shown in Fig. 4.2b (the
interaction distance is taken as 6.5 Å from the C-α coordinates), the contact energy
between the residues is given by each element of E for each type of residue in Eq. 4.1,
i.e., when residue i is H ( 0 1ix≤ < ) and residue j is H ( 0 1j
x≤ < ), energy of interaction
is HHe , when residue i is H ( 0 1ix≤ < ) and residue j is P (1 2jx≤ < ), energy of
interaction is HPe , and so on. A continuous function representation of the matrix in
Eq. 4.1 is shown in Fig. 4.2c and the mathematical formula is a two-dimensional sigmoid
function as shown below,
( )
( )( )( )
( )( )( )
( )( ) ( )( )
1 1
2
1 1
j jii
j jii
HP HHHHij x xc xx
HP HH PP HP HH
c x c xc xx
e eeE
e e e e
e e e e e
e e e e
α ααα
α ααα
− −−−
− −−−
−= + +
+ + + +
− − ++
+ + + +
(4.2)
where,
α = smoothening parameter of the sigmoid function
= 25
c = 1
In a similar manner, the two-dimensional sigmoid function for twenty amino acids can be
constructed. Thus, for 20 amino acids, the formula is given by,
( ) ( )( )20 20
111 1
( , )
1 ji
ijx jx i
j i
f i jE
e eαα − − +− − +
= =
=+ +
∑∑ (4.3)
where the function ( , )f i j is given by,
( , ) ( , ) ( , )f i j M i j g i j= + (4.4)
where,
( , )M i j = the ijth value of the inter-residue contact energy matrix
and ( , )g i j is obtained recursively as,
( )( ) ( )1 1
(1,1) (1,1)
( , )( , ) ( , ) 1, 1
1
ji
i k j mk m
g M
g k mg i j M i j i j
e eβ ε β ε− + − − + −
= =
=
= − > >+ +
∑∑ (4.5)
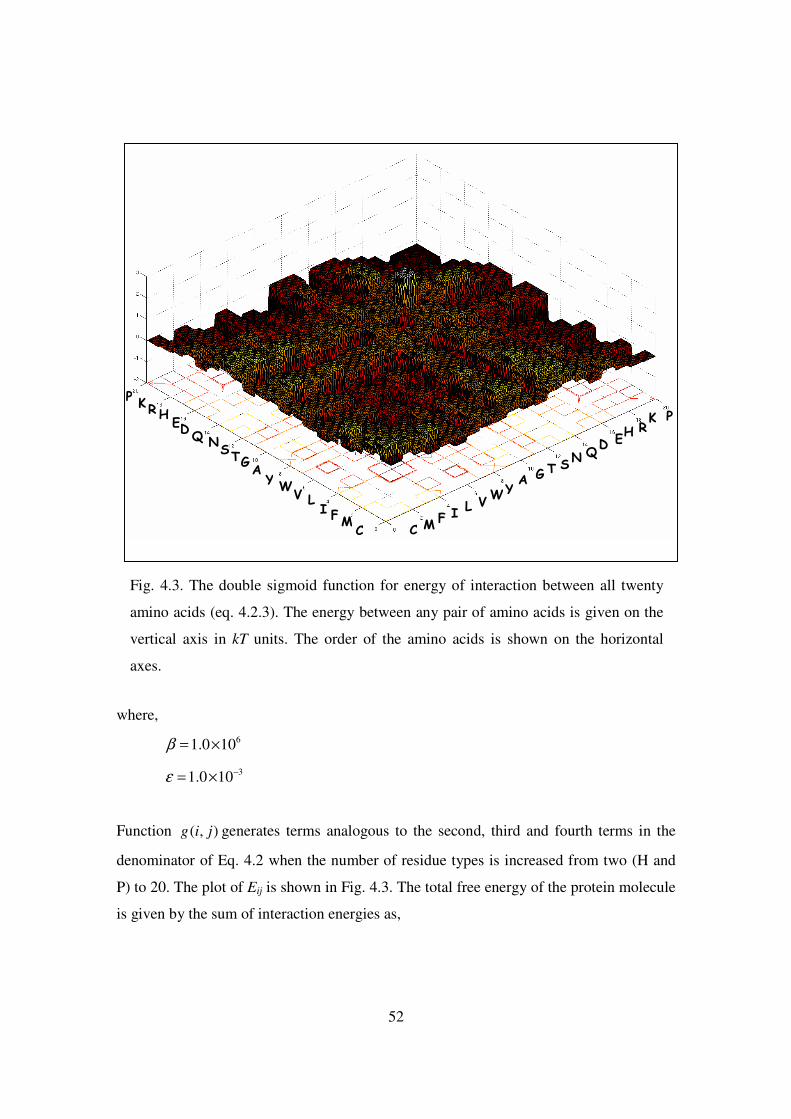
52
where,
61.0 10β = ×
31.0 10ε −= ×
Function ( , )g i j generates terms analogous to the second, third and fourth terms in the
denominator of Eq. 4.2 when the number of residue types is increased from two (H and
P) to 20. The plot of Eij is shown in Fig. 4.3. The total free energy of the protein molecule
is given by the sum of interaction energies as,
Fig. 4.3. The double sigmoid function for energy of interaction between all twenty
amino acids (eq. 4.2.3). The energy between any pair of amino acids is given on the
vertical axis in kT units. The order of the amino acids is shown on the horizontal
axes.
P
C M F I
L W Y A G T S
N Q
D E H R K
C M F I L
W Y A G T S
N Q
D E H R K
V V
P P

53
( )1 1
1( , ) ,
2i j
N N
Total i j ij i j
x x
E C x x E x x= =
= ∑∑ (4.6)
where,
( , )i jC x x = 1 if residues at positions ix and jx are within the interaction distance
= 0 elsewhere
and Eij is the energy of interaction between the residues of type i and j at positions ix and
jx respectively.
Apart from the inter-residue contact energy matrix, the ZSk potential also
contains two tables that give propensities of amino acids to form different secondary
structures, i.e., alpha helices and beta strands. These tables can be incorporated as
continuous functions in the following manner,
( )
20
/ ( 1)1
( )( )
1 x ii
h iE i
eα β α− − +
=
=+
∑ (4.7)
where / ( )E iα β is the secondary structure propensity energy (Fig. 4.4) for residue-type i
(alpha or beta depending on what type of secondary structure the residue lies in) and the
function ( )h i is given by,
( ) ( ) ( )h i S i p i= + (4.8)
where
( )S i = the secondary structure propensity value for residue-type i from Zhang
Skolnick’s secondary structure propensity table
and ( )p i is obtained recursively as
( )( )1
(1) (1)
( )( ) ( ) 1
1
i
i kk
p S
p kp i S i i
eβ ε− + −
=
=
= − >+
∑ (4.9)
where the parameters ,α β and ε have the same values as described before. Thus, the
total energy of the polypeptide when using the ZSk potential becomes,
( ) /1 1 1
1( , ) , ( )
2i j i
N N N
Total i j ij i j i
x x x
E C x x E x x E xα β= = =
= +∑∑ ∑ (4.10)
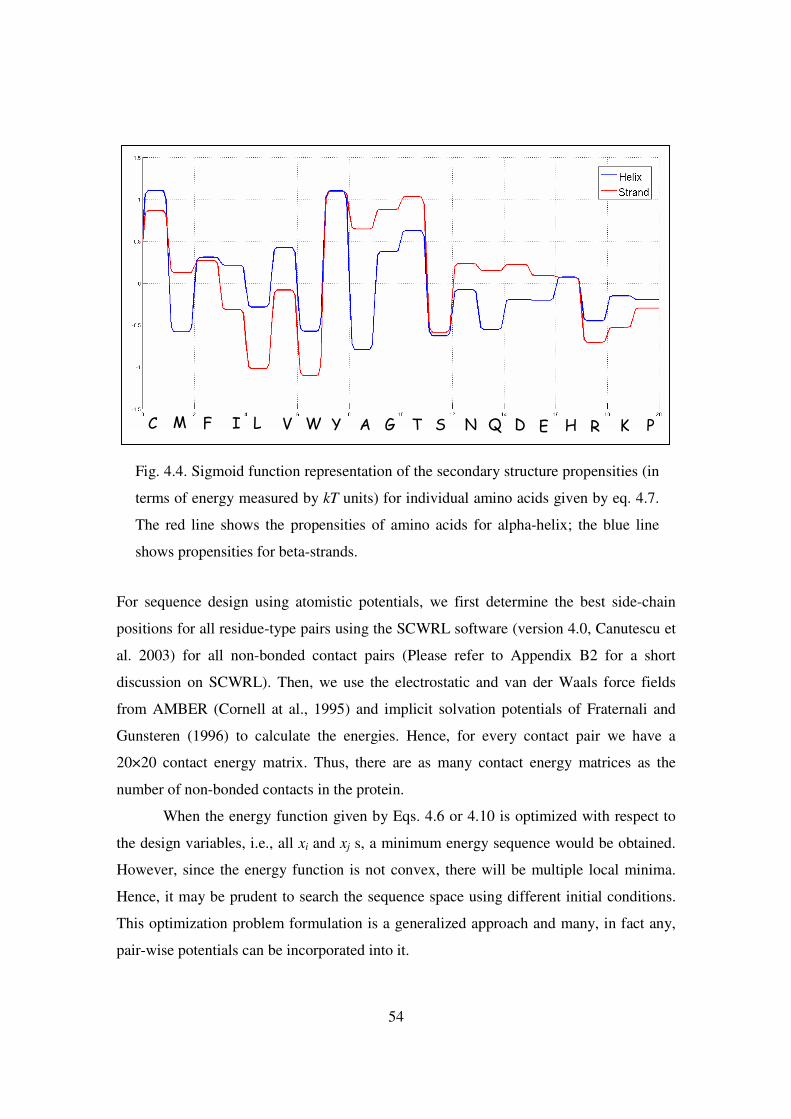
54
For sequence design using atomistic potentials, we first determine the best side-chain
positions for all residue-type pairs using the SCWRL software (version 4.0, Canutescu et
al. 2003) for all non-bonded contact pairs (Please refer to Appendix B2 for a short
discussion on SCWRL). Then, we use the electrostatic and van der Waals force fields
from AMBER (Cornell at al., 1995) and implicit solvation potentials of Fraternali and
Gunsteren (1996) to calculate the energies. Hence, for every contact pair we have a
20×20 contact energy matrix. Thus, there are as many contact energy matrices as the
number of non-bonded contacts in the protein.
When the energy function given by Eqs. 4.6 or 4.10 is optimized with respect to
the design variables, i.e., all xi and xj s, a minimum energy sequence would be obtained.
However, since the energy function is not convex, there will be multiple local minima.
Hence, it may be prudent to search the sequence space using different initial conditions.
This optimization problem formulation is a generalized approach and many, in fact any,
pair-wise potentials can be incorporated into it.
Fig. 4.4. Sigmoid function representation of the secondary structure propensities (in
terms of energy measured by kT units) for individual amino acids given by eq. 4.7.
The red line shows the propensities of amino acids for alpha-helix; the blue line
shows propensities for beta-strands.
C M F I L V W Y A G T S N Q D E H R K P

55
4.2.2 Formulation of the constraints
In cases when the amino acid composition of the target sequences are known, one may be
interested in designing sequences keeping the amino acid composition fixed. As proposed
by Koehl and Levitt (1999, 2002), this criterion ensures that the designed sequences are
specific to the target structure. In our optimization problem formulation, we can
incorporate the amino acid composition constraints. Let there be km numbers of residue
type k for each of the twenty types of amino acids. Then,
20
1k
k
m N=
=∑ (4.11)
where N is the total number of residues in the protein. km is related to the design
variables by the equation,
( )( ) ( )( )1 2
1
1 1
1 1i i
N
kx c x ci
me e
α α− −=
− =
+ + ∑ (4.12)
where 1c and 2c are the upper and lower ranges for residue type k, i.e., for example,
1c = 0.0 and 2c = 1.0 for Cystine from Fig. 4.3 and α is the smoothness parameter for the
sigmoid function. The plot of constraints against each residue position is shown in Fig.
4.5.
We would like to mention here that due to incorporation of such nonlinear
constraints (one constraint for each type of residue), often, optimization doesn’t
converge.
4.2.3 Results
As mentioned before, we choose a set of four proteins (PDB IDs 1FC2 chain C, 1HDD
chain C, 2CRO and 4ICB) to test our method. Even though we design sequences solely
based on minimization of the energy as given by Eq. 4.6 or 4.10, we thread the designed
sequences on the decoy set structures and check for specificity by calculating the energy
gap between the native structure and other structures in the decoy set and also standard
deviation of sequence energies over the structures in the same decoy set. Based on the
design criteria, i.e., minimization of energy and different initial values of the design
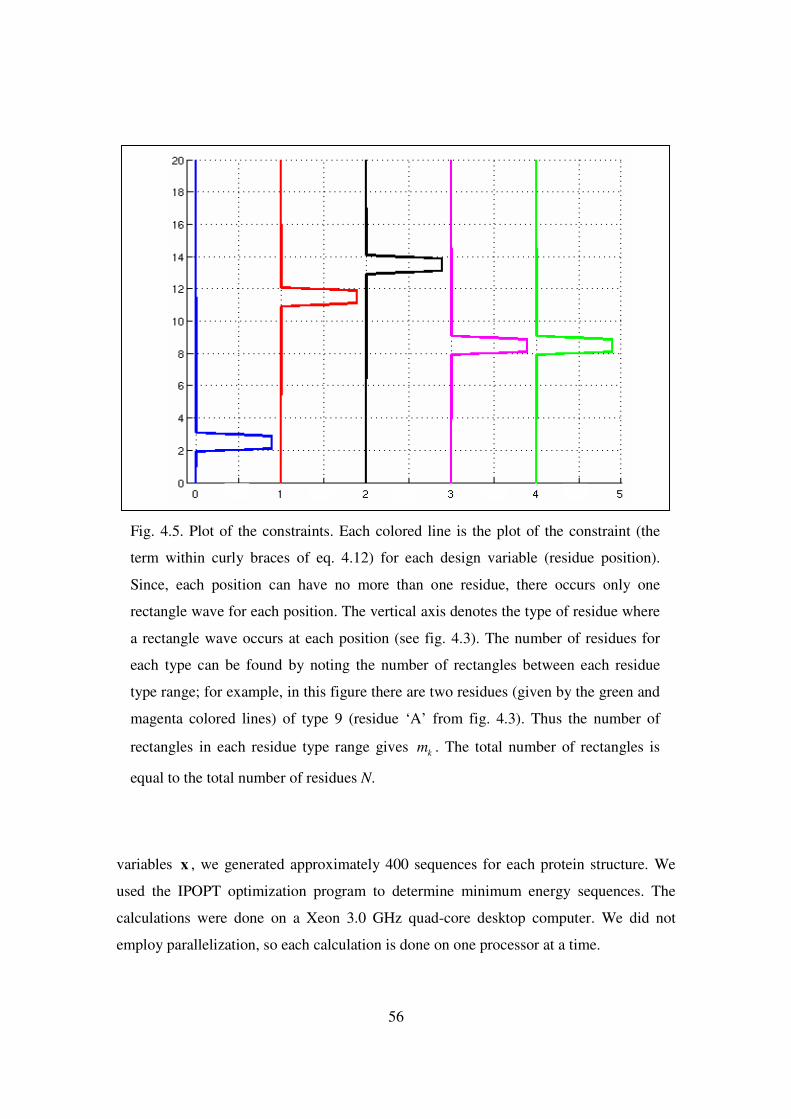
56
variables x , we generated approximately 400 sequences for each protein structure. We
used the IPOPT optimization program to determine minimum energy sequences. The
calculations were done on a Xeon 3.0 GHz quad-core desktop computer. We did not
employ parallelization, so each calculation is done on one processor at a time.
Fig. 4.5. Plot of the constraints. Each colored line is the plot of the constraint (the
term within curly braces of eq. 4.12) for each design variable (residue position).
Since, each position can have no more than one residue, there occurs only one
rectangle wave for each position. The vertical axis denotes the type of residue where
a rectangle wave occurs at each position (see fig. 4.3). The number of residues for
each type can be found by noting the number of rectangles between each residue
type range; for example, in this figure there are two residues (given by the green and
magenta colored lines) of type 9 (residue ‘A’ from fig. 4.3). Thus the number of
rectangles in each residue type range gives km . The total number of rectangles is
equal to the total number of residues N.

57
To demonstrate the generality of our approach, we use three different potentials for
energy calculation. As mentioned before, these are the Miyazawa Jernigan (MJ), Zhang
Skolnick (ZSk), and the atomistic potentials based on electrostatic and van der Waal’s
parameter values from the AMBER force field and solvation energy from implicit
solvation model of Fraternali and Gunsteren. For each protein, we design sequences both
with and without amino acid composition constraints. After designing the sequences we
align them with the corresponding wild-type sequence of each protein. For finding the
best sequence based on alignment, we not only calculate the number of amino acid
matches but also the matches based on the reduced amino acid sets. Apart from
hydrophobic-hydrophilic grouping of amino acids, we find grouping amino acids into
five sets most common in the literature. Here, we select 10 different five-letter grouping
schemes (Dayhoff et al. 1978, Wang and Wang 1999, Murphy et al. 2000, Cieplak et al.
2001, Cannata et al. 2002, Wang and Wang 2002, Li et al. 2003, Koisol et al. 2004,
Rakshit and Ananthasuresh 2008) based on different criteria and calculate matches
between designed and wild-type sequences after aligning them. Each grouping scheme
uses its own method for reducing the amino acid set and brings out common
characteristics between different amino acids based on which they are grouped. For
example, whereas Dayhoff’s classification (1978) is based on evolutionary criteria and
calculated using the PAM matrix, Rakshit and Ananthasuresh’s classification (2008) is
based on hydrophobicity-hydrophilicity criterion and calculated using the MJ matrix. We
calculate the total number of reduced-alphabet matches for each designed sequence based
on all grouping methods and add the number of non-reduced matches after weighting
them by a factor of four. This factor is introduced to compensate for the loss of
specificity from twenty to five letters. We can thus rank the designed sequences based on
a score given by

58

59
60
Table 4.1. Number of matches of the best designed sequences given in Fig. 4.6 using
different grouping schemes for each of the four proteins using all three types of
potentials. The first row for each of the proteins denoted by “All twenty” indicates
number of matches where no grouping schemes have been used. Wang* and Wang**
implies their grouping schemes used in (1999) and (2002) works respectively. For
brevity, we denote each grouping scheme by the name of the first authors only.
MJ ZSk Atomistic Protein
PDB
ID and
number
of
residue
-s
Groupin
-g
scheme
Amino acid
compositio
-n not
conserved
Amino acid
compositio
-n
conserved
Amino acid
compositio
-n not
conserved
Amino acid
compositio
-n
conserved
Amino acid
compositio
-n not
conserved
All
twenty 9 9 10 7 8
Dayhoff 24 22 22 22 20
Wang* 15 14 18 14 14
Murphy 17 17 16 20 10
Cieplak 21 20 21 20 17
Cannata 21 20 23 25 17
Wang** 15 14 20 16 13
1FC2
(chain
C)
43
Li 18 19 21 22 17
Fig. 4.6. Best designed sequences based on our scoring scheme using different
potentials with and without amino acid composition constraints for each of the four
proteins. We also show the results of sequence alignment using sequence alignment
program CLUSTAL (version 1.83). ‘*’ denotes a match; ‘:’ denotes conserved
substitution; ‘.’ denotes semi-conserved substitution.
(http://www.ebi.ac.uk/help/formats . html )

61
Koisol 27 27 28 27 25
Rakshit 18 15 20 16 16
All
twenty 12 9 10 8 9
Dayhoff 23 17 26 17 28
Wang* 20 19 23 20 27
Murphy 16 13 21 10 23
Cieplak 26 24 27 24 28
Cannata 26 20 30 23 31
Wang** 20 17 21 17 22
Li 23 20 28 19 29
Koisol 34 32 37 32 39
1 HDD
(chain
C)
57
Rakshit 21 15 23 14 22
All
twenty 11 11 10 14 10
Dayhoff 26 28 29 29 24
Wang* 24 19 29 25 22
Murphy 18 21 24 22 18
Cieplak 26 22 24 26 21
Cannata 26 27 33 23 25
Wang** 20 16 21 22 20
Li 23 24 29 20 23
Koisol 35 35 38 38 33
2 CRO
65
Rakshit 26 24 28 30 23
All
twenty 16 14 14 13 12
Dayhoff 36 34 37 34 33
Wang* 32 24 26 21 25
Murphy 25 24 26 24 22
4 ICB
76
Cieplak 38 34 38 29 30
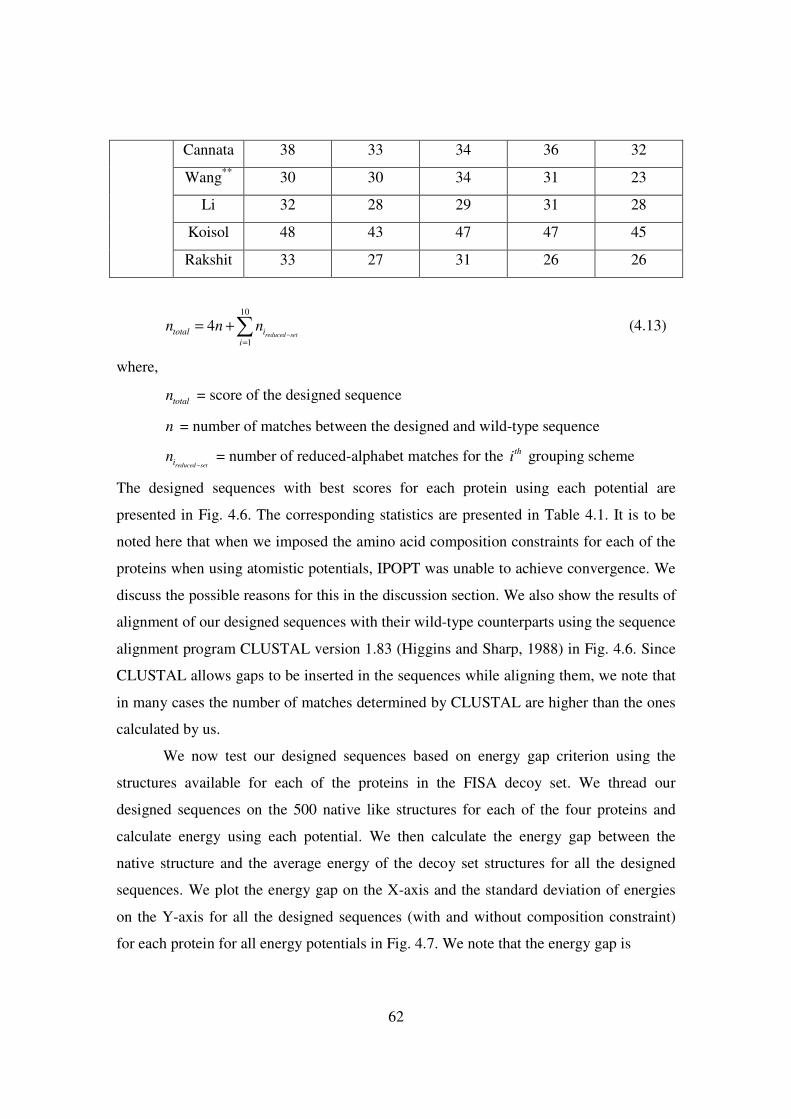
62
Cannata 38 33 34 36 32
Wang** 30 30 34 31 23
Li 32 28 29 31 28
Koisol 48 43 47 47 45
Rakshit 33 27 31 26 26
10
1
4reduced settotal i
i
n n n−
=
= +∑ (4.13)
where,
totaln = score of the designed sequence
n = number of matches between the designed and wild-type sequence
reduced setin
− = number of reduced-alphabet matches for the th
i grouping scheme
The designed sequences with best scores for each protein using each potential are
presented in Fig. 4.6. The corresponding statistics are presented in Table 4.1. It is to be
noted here that when we imposed the amino acid composition constraints for each of the
proteins when using atomistic potentials, IPOPT was unable to achieve convergence. We
discuss the possible reasons for this in the discussion section. We also show the results of
alignment of our designed sequences with their wild-type counterparts using the sequence
alignment program CLUSTAL version 1.83 (Higgins and Sharp, 1988) in Fig. 4.6. Since
CLUSTAL allows gaps to be inserted in the sequences while aligning them, we note that
in many cases the number of matches determined by CLUSTAL are higher than the ones
calculated by us.
We now test our designed sequences based on energy gap criterion using the
structures available for each of the proteins in the FISA decoy set. We thread our
designed sequences on the 500 native like structures for each of the four proteins and
calculate energy using each potential. We then calculate the energy gap between the
native structure and the average energy of the decoy set structures for all the designed
sequences. We plot the energy gap on the X-axis and the standard deviation of energies
on the Y-axis for all the designed sequences (with and without composition constraint)
for each protein for all energy potentials in Fig. 4.7. We note that the energy gap is

63
1FC2
a(i)
a(ii)
a(iii)

64
b(i)
1HDD
b(ii)
b(iii)
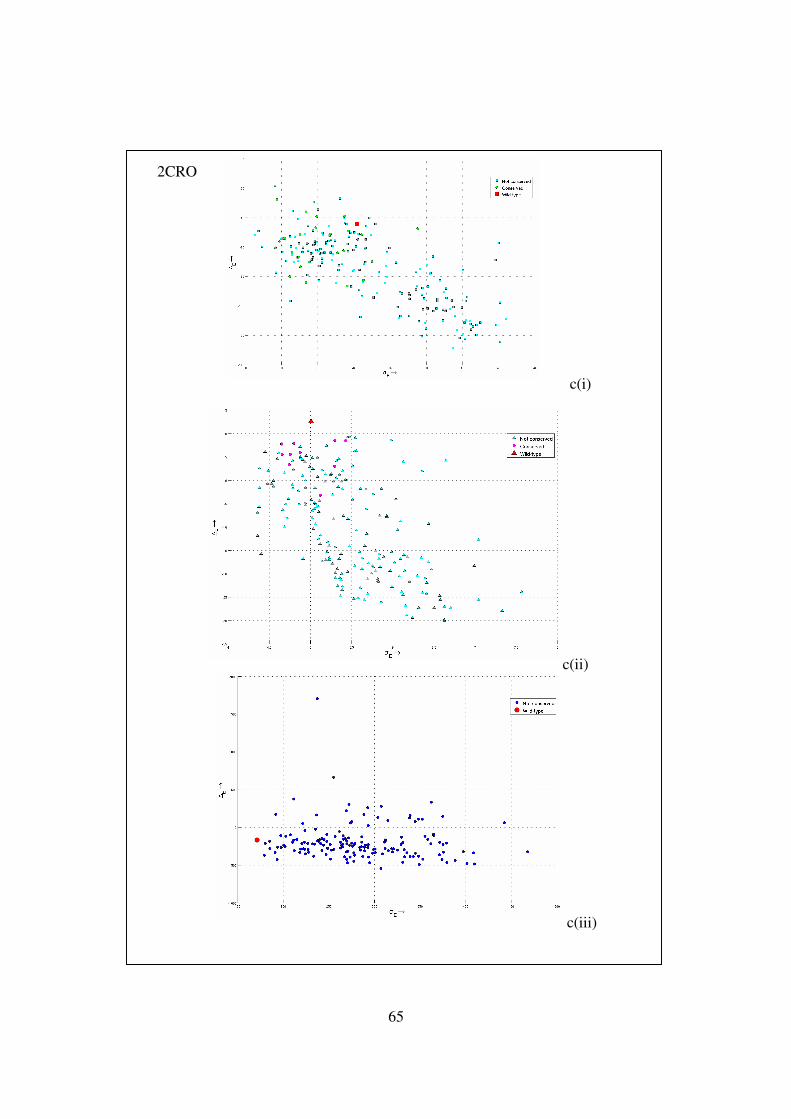
65
2CRO
c(i)
c(ii)
c(iii)

66
4ICB
d(i)
d(ii)
d(iii)

67
Fig. 4.7. Plots of energy gap (E native avg decoys
E E −∆ = − ) versus Eσ (the standard
deviation of energy of the decoy set structures). The name of the protein is indicated
on top left of each figure. For all the proteins, the first plot is for MJ potential, the
second for ZSk potential and the third for atomistic potential.
a(i). E∆ versus Eσ for 1FC2 using MJ potential. The red square indicates the result
for the wild-type sequence. The cyan squares represent designed sequences for
which amino acid composition constraints was not imposed. The green dots
represent designed sequences for which amino acid composition constraints were
satisfied. The legend is indicated on top right of the figure.
a(ii). E∆ versus Eσ for 1FC2 using ZSk potential. The red triangle indicates the
result for the wild-type sequence. The cyan triangles represent designed sequences
for which amino acid composition constraints was not imposed. The magenta dots
represent designed sequences for which amino acid composition constraints were
satisfied. The legend is indicated on top right of the figure.
a(iii). E∆ versus Eσ for 1FC2 using atomistic potential. The red dot indicates the
result for the wild-type sequence. The blue dots represent designed sequences for
which amino acid composition constraints was not imposed. The legend is indicated
on top right of the figure.
b(i) E∆ versus Eσ for 1HDD using MJ potential. Color code is same as that of a(i).
b(ii) E∆ versus
Eσ for 1HDD using ZSk potential. Color code is same as that of
a(ii). b(iii) E∆ versus Eσ for 1HDD using atomistic potential. Color code is same as
that of a(iii).
c(i) E∆ versus Eσ for 2CRO using MJ potential. Color code is same as that of a(i).
c(ii) E∆ versus Eσ for 2CRO using ZSk potential. Color code is same as that of
a(ii). c(iii) E∆ versus
Eσ for 2CRO using atomistic potential. Color code is same as
that of a(iii).
d(i) E∆ versus
Eσ for 4ICB using MJ potential. Color code is same as that of a(i).
d(ii) E∆ versus Eσ for 4ICB using ZSk potential. Color code is same as that of a(ii).
d(iii) E∆ versus
Eσ for 4ICB using atomistic potential. Color code is same as that of
a(iii).

68
always negative, i.e., the native state energy is less than the average of the decoy
structure energies for all the four proteins when we use the atomistic potential. This
implies that the wild-type sequence will favorably select the native structure over other
native-like structures. However, when we use the other potentials, we note that the energy
gap is not always negative for all the four proteins. We got negative energy gap for only
4ICB with the MJ potential, while negative energy gap was achieved for only 1FC2 with
ZSk potential. Thus, even though we are able to achieve convergence by keeping the
amino acid composition fixed for ensuring specificity while using MJ and ZSk potentials,
but are unable to do so while using atomistic potentials, the sequences designed using
atomistic potentials seem to be more appropriate as they satisfy the negative energy-gap
criterion. It is interesting to note that many of our designed sequences have energy gap
lower than that of the corresponding wild-type sequence. Some may find it surprising but
it is not uncommon in de novo protein design literature to come across works where
sequences have been redesigned to be more stable than their wild-type counterparts
(Chen et al., 2000, Dantas et al., 2003).
We also present the average time taken by IPOPT for designing sequences using
different potentials for all the four proteins in Table 4.2. Even though gradient-based
continuous optimization methods are computationally efficient in determining a local
minimum, the average time taken by IPOPT for designing sequences for the four proteins
(especially using atomistic potentials) is quite high in our opinion. In the next section, we
present the quadratic programming approach for designing sequences which is much
faster.

69
Table 4.2. Average time taken for designing sequences of each protein using MJ
(Miyazawa and Jernigan, 1996), ZSk (Zhang and Skolnick, 1998) and atomistic
potentials (Cornell at al., 1995, Fraternali and Gunsteren, 1996).
Protein PDB ID
and number of
residues
MJ ZSk Atomistic
1FC2 (chain C)
43 30 m 8 s 1 h 19 m 32 s 20 m 51 s
1HDD (chain C)
57 1h 36 m 2 s 2 h 55 s 3 h 36 m 2 s
2CRO
65 2 h 1 m 14 s 2 h 13 m 30 s 5 h 11 m 15 s
4ICB
76 3 h 12 m 10 s 2 h 51 m 21 s 6 h 4 m 58 s
4.3 The Quadratic Programming method
4.3.1 Method
In the quadratic programming approach, for an N residue protein we select a vector
20N∈x � with each element representing a particular amino acid at a particular position
in the sequence. Thus, at the thi residue position in the sequence, the th
m type of residue
will be identified by the element 20( 1)i mx − + of x . Hence, the dimension of x is 20 1N × .
The following ordering was used for the amino acids: {C M F I L V W Y A G T S N Q D
E H R K P}. The sequence design problem is formulated, following Koh et al. (2005) as
follows,
1Minimize
2T
E =x
x Qx (4.14)
Subject to =Bx c (4.15)
and 0 1 1, 20ix i N≤ ≤ ∀ =
where zero for an entry in x means vacancy at a corresponding site by a corresponding
amino acid residue and one implies occupancy at the same site by that particular residue.

70
Q is the energy matrix; the thij entry of Q gives the contact energy between the thi and
thj residues when the Cα atoms are within a contact distance of 7.0 Å of each other. The
contact energy consists of three atomistic potentials, namely, electrostatic, van der Waals
and solvation potentials. The atomistic electrostatic charges and van der Waals
parameters are taken from the AMBER force field (Cornell et al., 1995) and for solvation
energy, we use the implicit solvation energy model proposed by Fraternali and Gunsteren
(1996). The contact energy depends on the orientations of the side chains of the residues
in contact. We use SCWRL version 4 (2003) to determine the orientation of the side-
chains. SCWRL uses the backbone coordinates of each pair of interacting residues in the
native structure and gives the best side-chain orientation.
Based on assumptions stated above, the pair-wise contact energy is given by,
ij electrostatic VW solvation
Q E E E= + + if the thi and thj residues are in contact
= 0 otherwise
Thus, after expansion, Eq. 4.14 takes the following form
20 20
20( 1) 20( 1) ,20( 1) 20( 1)1 1 1 1
12
N N
i k i k j l j l
i j k l
E x Q x− + − + − + − += = = =
=
∑∑ ∑∑ (4.16)
where the indices i and j indicate the positions on the backbone that are in contact, and
the indices k and l indicate the type of amino acid at positions i and j respectively. B is
the constraint matrix and c the constraint vector. The first N rows of B and c indicate
the number of residues at any position on the backbone. Since, only one amino acid can
occupy a position on the backbone at a time, the first N rows of c are all ones. The thi
row of B ( i N≤ ) gives the coefficients of the variables { }x corresponding to the thi
position on the backbone; the only variables active at the thi position are from
20( 1) 1i − + to 20i . Thus for row i,
1ij
B = for 20( 1) 1 to 20j i i= − +
= 0 otherwise
Hence, the first N rows of the constraint eq. 4.4.2 can be written as
20
20( 1)1
1 for 1i j
j
x i N− +=
= ≤ ≤∑ (4.17)

71
The last 20 rows of B and c specify the amino acid composition of the sequence to be
designed. Thus, if the sequence to be designed should have five Glycines, then the
( )10th
N + row of c is equal to five (note that in the order of amino acids mentioned
above Glycine is at the 10th position). On the other hand, in the ( )10th
N + row of the left
hand side of Eq. 4.15, only variables corresponding to Glycine should be present. Hence,
every 10th element of B in the 10 thN + row should be one while the rest are zeroes.
Thus, the last 20 rows of the constraint Eq. 4.15 can be written as
20( 1)1
for 1 20N
j m m
j
x N m− +=
= ≤ ≤∑ (4.18)
where mN is the number of amino acids of type m. We also ensure that,
20
1m
m
N N=
=∑ (4.19)
because the sum of the number of different types of amino acids should add up to the
length of the sequence. From Eqs. 4.17 and 4.18 it is easy to see that the dimension of B
is (20 ) 20N N+ × and that of c is (20 ) 1N+ × .
We solve the optimization problem posed in Eqs. 4.14 and 4.15 by using the
interior point optimization method (IPOPT) (Wächter and Biegler, 2004) (refer Appendix
B1). Interior point optimization methods are efficient in handling a large number of
optimization variables, nonlinear objective function, and a large number of constraints.
As an example, for designing the smallest protein sequence which has 46 residues we
have 46 20 920× = design variables and 46 20 66+ = constraints along with the
optimization function given by Eq. 4.16. For such problems, IPOPT takes only six to
seven minutes on a Xeon 3.0 GHz desktop computer.
4.3.2 Results
Optimization using quadratic programming formulation is much faster compared to the
double sigmoid approach. We used quadratic programming on the same set of four
proteins in the FISA decoy set to design sequences. Based on these design criteria and
different initial values of the design variables x , we generate approximately 100
sequences for each protein structure. Table 4.3 gives the average time required for

72
Table 4.3 Average time taken to design sequences for each protein in the FISA decoy set
using quadratic programming formulation.
PDB ID Number of residues Average time
1FC2 (chain C) 43 7 m 39 s
1HDD (chain C) 57 16 m 47 s
2CRO 65 24 m 10.5 s
4ICB 76 46 m 30.5 s
designing set of sequences for each protein using quadratic programming formulation.
All the calculations were done in a Xeon 3.0 GHz quad-core desktop computer. As
before, we did not employ parallelization, so each calculation was done on one processor
at a time.
As explained in Section 4.3, we align the designed sequences with the wild-type
ones and also find matches with reduced set of five amino acid alphabets using the same
grouping schemes mentioned in section 4.3. We use the scoring scheme in Eq. 4.13 to
select the best designed sequences and present them in Fig. 4.8. The reduced amino acids
are represented by ‘A’, ‘B’, ‘C’, ‘D’ and ‘E’. When we align our maximum score
sequences with their wild-type counterparts by using the sequence alignment program
CLUSTAL version 1.83 (Higgins and Sharp, 1988) which allows gaps to be introduced in
the sequences, we notice that the number of matches increase. The results of CLUSTAL
alignment for the four proteins are shown in Fig. 4.9. As explained in Section 4.3, we
again test our designed sequences by threading them to the decoy structures in the FISA
decoy set and note the energy gaps and energy dispersions for these four proteins. The
results are presented in Fig. 4.10.

73
(a)

74
(b)

75
(c)

76
(d)

77
(a)
(b)
(d)
(c)
Fig. 4.8. The highest scoring designed sequences for the four proteins 1FC2 (chain
C), 1HDD (chain C), 2CRO and 4ICB. Each match is shown by a ‘|’ symbol. For
each protein, we first show the number of matches with all 20 amino acids which we
term as ‘exact match’, and then each five letter reduced amino acid sets denoted by
the corresponding authors. For every case, the top is the wild-type and the bottom
one is the designed sequence. The number of matches for each method is shown
below the each grouping scheme as well as the non-reduced one. Wang&Wang1,
Wang&Wang2 and Wand&Wang2002 refers (Wang and Wang, 1999 and Wang and
Wang, 2002) grouping schemes respectively.
(a) 1FC2 (chain C); 18.6 % amino acid matches without introducing gaps.
(b) 1HDD (chain C); 17.5 % amino acid matches without introducing gaps.
(c) 2CRO; 13.8 % amino acid matches without introducing gaps.
(d) 4ICB; 14.5 % amino acid matches without introducing gaps.

78
In Fig. 4.10, we note that energy gap, i.e., E native avg decoysE E −∆ = − is negative for all the
designed sequences (shown as blue stars) as well for the wild-type sequence (shown as a
red dot). Since, the free energy is always negative in sign, all our designed sequences as
well as the wild-type sequences satisfy the first criteria, i.e., the energy of the native
structure is lower than that of the decoy set structures. However, we see that the
dispersion in energy of the decoy structures is almost similar in magnitude as the energy
gap for 1FC2-C, 1HDD-C and 2CRO for the wild-type as well as the designed sequences.
Based on the definition of the Z-score (Shakhnovich and Gutin, 1993, Abkevich and
Shakhnovich, 1996) , i.e.,
_
_
decoy setnative
decoy set
E EZ
σ
−= (4.20)
we see that the dispersion in energy of the decoy structures is almost similar in magnitude
as the energy gap for 1FC2-C, 1HDD-C and 2CRO for the wild-type as well as the
designed sequences. Thus, for these three proteins 1Z ≈ , which is not encouraging from
the Z-score optimization point of view. This leaves the possibilities for improvement in
energy models and the implementation scheme which we discuss later. However, for the
other protein, 4ICB, we note that 4Z > for many designed sequences as well as for the
wild-type sequence. Thus, the designed sequences for this protein perfectly satisfies the
Z-score optimization criteria even though our sequence design method is not based on Z-
score optimization. As before, we note that some of the designed sequences are more
stable than the wild-type sequence based on the Z-score maximization criterion.
Fig. 4.9. Results of sequence alignment using sequence alignment program CLUSTAL
(version 1.83). ‘*’ denotes a match; ‘:’ denotes conserved substitution; ‘.’ denotes semi-
conserved substitution. ( http://www.ebi.ac.uk/help/formats.html ). As indicated, the top
sequence is the wild-type variety and the bottom the designed one.
(a) 1FC2 (b) 1HDD-C (c) 2CRO and (d) 4ICB.
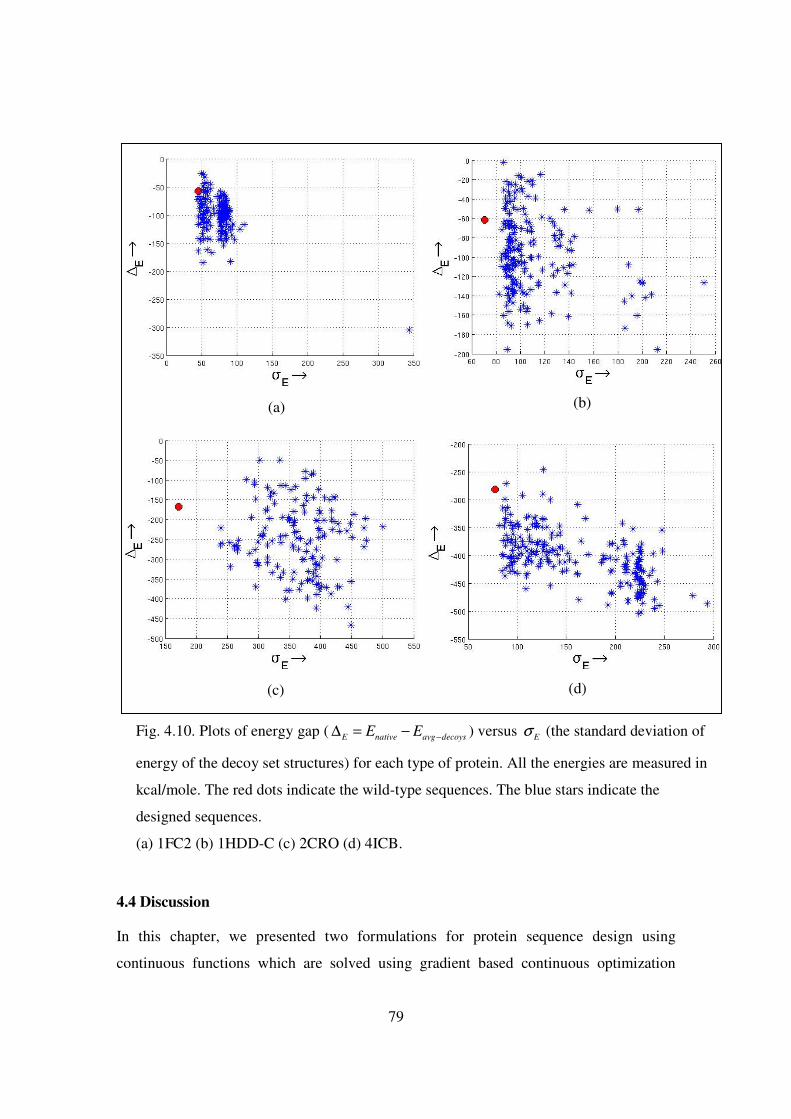
79
4.4 Discussion
In this chapter, we presented two formulations for protein sequence design using
continuous functions which are solved using gradient based continuous optimization
Fig. 4.10. Plots of energy gap ( E native avg decoysE E −∆ = − ) versus Eσ (the standard deviation of
energy of the decoy set structures) for each type of protein. All the energies are measured in
kcal/mole. The red dots indicate the wild-type sequences. The blue stars indicate the
designed sequences.
(a) 1FC2 (b) 1HDD-C (c) 2CRO (d) 4ICB.
(a) (b)
(c) (d)

80
methods. We now discuss a few merits and demerits of both the methods and also about
sequence design by free energy minimization using pair-wise contact potentials.
The double sigmoid method takes considerably more time (Table 4.2) than the
quadratic programming method (Table 4.3). Furthermore, with amino acid composition
constraints, especially with the atomistic potentials, the double sigmoid method was
unable to converge in many instances. On the other hand, the quadratic programming
method always converged with amino acid composition constraints using atomistic
potentials with different initial guesses. There is another inherent disadvantage of the
double sigmoid method; because of the nonconvexity on the energy surface it explores
(see Fig. 4.3), the solutions found by this method will depend on the contour of the
energy surface. Thus, if the energy surface profile is changed, which can be done by
altering the positions of amino acids, i.e., shifting rows and columns of the energy
matrices, the solutions found by the double sigmoid method will change for the same
initial inputs. Hence, the double sigmoid method is dependant on the order in which the
amino acids are presented in the energy matrices. As each amino acid represents a
separate design variable in the quadratic programming problem, no such problem will be
encountered. On the other hand, any value of the design variable between zero and
twenty represents an amino acid in the double sigmoid method; hence, whenever it
converges, it gives a specific sequence. However, in the quadratic programming
formulation, a specific amino acid is obtained only if the design variable corresponding to
it is one or near one (in our computer programs we kept this limit as greater than or equal
to 0.9). Hence, even though the quadratic programming method may converge obeying
all constraints, the final answer may not correspond to a protein sequence.
The present formulation based on pair-wise contact energies has a drawback when
we use atomistic potentials with implicit solvation model. Pair-wise energy calculation is
valid for electrostatic and van der Waals energies, but solvation energy depends upon
exposed surface area of the residues which is not pair-wise additive. Thus, the surface
area of a group of residues in close contact with one another is not equal to the sum of the
surface areas when two residues are taken at a time; in fact this leads to over-counting of
the surface area (Gordon and Mayo, 1999). One has to take all the atoms of all the
residues in contact simultaneously to calculate the surface area correctly. However,

81
determining the correct solvation energy by taking all residues together will lead to a high
increase in the computational cost. For example, if m residues are in contact with one
another, we now have 2
m
(i.e., ( )
!2 !2!
m
m −) contact energy matrices of size 20×20.
However, if we want to calculate all possible combinations of contact energies of 20
amino acids for such a m residue group we will have to calculate a 20×20×20×….×20
contact energy matrix of m dimensions which leads to an exponential increase in
dimensionality. The energy evaluation formula for the quadratic programming method
(eq. 4.4.3) will then be modified as,
1 1 2 2 1 2 2 2
1 2 1 2
20 20 20
20( 1) ,20( 1) ,....20( 1) 20( 1) 20( 1) 20( 1)1 1 1 1 1 1
1.... .... ....
2 m m m m
m m
N N N
m i k i k i k i k i k i k
i i i k k k
E Q x x x− + − + − + − + − + − += = = = = =
=
∑∑ ∑ ∑∑ ∑
for every m group of residues taken together, and there may be several such closely
packed groups in a protein.
4.5 Closure
In this chapter we presented two approaches for protein sequence design for fixed
backbone conformations. Both the methods minimize free energy to design sequences
which is posed as a continuous function and solved using gradient based optimization
methods. We also developed formulations to impose amino acid composition constraints
in the continuous optimization problem framework. We demonstrated the generality of
the methods by incorporating different potentials ranging from coarse-grained statistical
ones to atomistic, and use both the methods to design sequences for four proteins of
varying chain lengths. We further tested the designed sequences by matching them with
the wild-type ones and also checked for stability by calculating the energy gap and
dispersion in energies using the decoy sets available for the four proteins. We end by a
discussion on the merits and demerits of each method.

82
5. Search in the Conformation Space
• We explain our approach towards protein structure prediction from the point of its
application in simultaneous sequence and conformation search.
• We describe the formulation of a coarse-grained energy function from the MJ
matrix.
• We present a structure prediction formulation based on elastic network model and
show its application by taking examples of real proteins.
• We formulate a continuous coarse-grained energy function to form coarse-grained
models of secondary structures like alpha helix.
• We describe a coarse-grained tertiary structure prediction method with rigid
secondary structures and validate it using several proteins.
• We present a discussion on both the structure prediction methods and select one
of them for future use in simultaneous sequence and conformation search
application.
• We close the chapter by a brief summary.
5.1 Introduction
The work presented in this chapter is concerned with the search of the conformation
space of the designed sequences for minimum-energy conformations. Even though the
conformation of the protein molecule has to obey constraints within the limits of the
Ramachandran map, still the protein molecule can take an infinite number of
conformations in the three-dimensional space. Consequently, searching the
conformational space is a computational intensive task. Hence, our focus is on techniques
that are amenable for computationally efficient energy potentials. First, we present a
novel coarse-grained continuous energy function based on the MJ matrix (Miyazawa and
Jernigan, 1996). Then, we present two conformation search methods, both of which use
the OB-CG (One-Bead Coarse-Grained) model that we developed. The first conformation
search method that we developed is based on the elastic network model, which we
implemented in MATLAB. This model is applied on a few proteins that show
preliminary results starting from fully unfolded states of polypeptides. However, we have

83
not used it subsequently because of some issues which we discuss in the relevant section
on discussions about this method. We have tried to predict secondary structure formation
using continuous optimization and have been successful to formulate a continuous
function which can form OB-CG model of alpha-helices when optimized starting from a
fully unfolded state. We present a short section on the description of this method.
However, alpha helix formation does not take place when we incorporate this function
with other terms in the OB-CG model. Henceforth, we developed a simple chain/linkage
model that incorporates secondary structures such as alpha helices and beta strands as
rigid bodies. However, the formation of beta sheets by pairing of beta strands is a
combinatorial problem and is still open and is not addressed using gradient based
continuous optimization. The chain/linkage model with pre-specified rigid secondary
structures works well with alpha-helical proteins in predicting tertiary structures from
unfolded states. To increase the computational efficiency, we developed our own
nonlinear conjugate gradient program and implemented the chain/linkage model using
C++ language (Please refer to Appendix B3 to see the algorithm). The results of this
method are given for some alpha helical proteins of different chain lengths. We also
present a case where this method is used for ab intio structure prediction with sequence
information only. We have used this method in our simultaneous sequence and
conformation search method presented in Chapter 6 of this thesis.
5.2 Coarse-grained energy function formulation
We use the latest MJ matrix (Miyazawa and Jernigan, 1996) to get the energy of
interaction between two non-bonded but interacting residues. Two non-bonded residues
are said to be interacting when they are within a distance of 6.5 Å from each other.
Since our model is based on MJ matrix, we want to make our energy model closely
follow the limits of the MJ matrix. Thus, the contact energy ijE between the thi and thj
non-bonded interacting residues in our coarse-grained model should be equal to the value
in the MJ matrix for the corresponding pair of residues at thi and thj positions. We have
modeled this using a sigmoid function which is the 1st term on the right hand side of Eq.
5.1. However, this does not prevent two residues from coming too close and even
overlapping on one another. To prevent this unrealistic scenario, we add a parabolic

84
function which comes into effect when two residues get closer than the sum of their radii
and is zero elsewhere. This is the 2nd term on right hand side of equation 5.1.
Thus, the energy of interaction between thi and thj non-bonded residues is given by,
ijE = ( )
2
1 exp 11 exp 1
ij cut inij
ijij
cut incut off
L Le
LL
LLββ
−
−−
−+
+ − −+ − −
(5.1)
where,
ije = contact energy between thi and thj residues given by the MJ matrix.
ijL = distance between thi and thj residues = ( ) ( ) ( )
2 2 2
i j i j i jx x y y z z− + − + −
cut inL − = cut-in distance; beyond this distance two non bonded residues repel one
another.
cut offL − = cut-off distance; beyond this distance MJ contact potential ije between
two residues cease to exist. The MJ contact potentials have been
calculated when two non bonded residues are within 6.5 Å of each
other. Thus in our case cut-off distance is cut inL − + 6.5 Å.
β = smoothness parameter = 15.
Figure 5.1 shows how the contact potential ijE between two non-bonded residues varies
with the distance ijL between them. One may note that our contact potential has a finite
value even when the centers of the two residues coincide with one another, which is
unrealistic. Even though this does not affect the calculations, it is physically possible to
overlap the centers of coarse-grained beads representing the centroid of the amino acid
residues without actual overlap of atoms constituting the residues.
We also have to take into account the constraints that restrict the conformational
space of proteins. Figure 5.2 shows how a 3sp -hybridized Cα atom restricts the
movement of its neighboring Cα atoms. We model this by providing a penalty term in
our energy formulation.
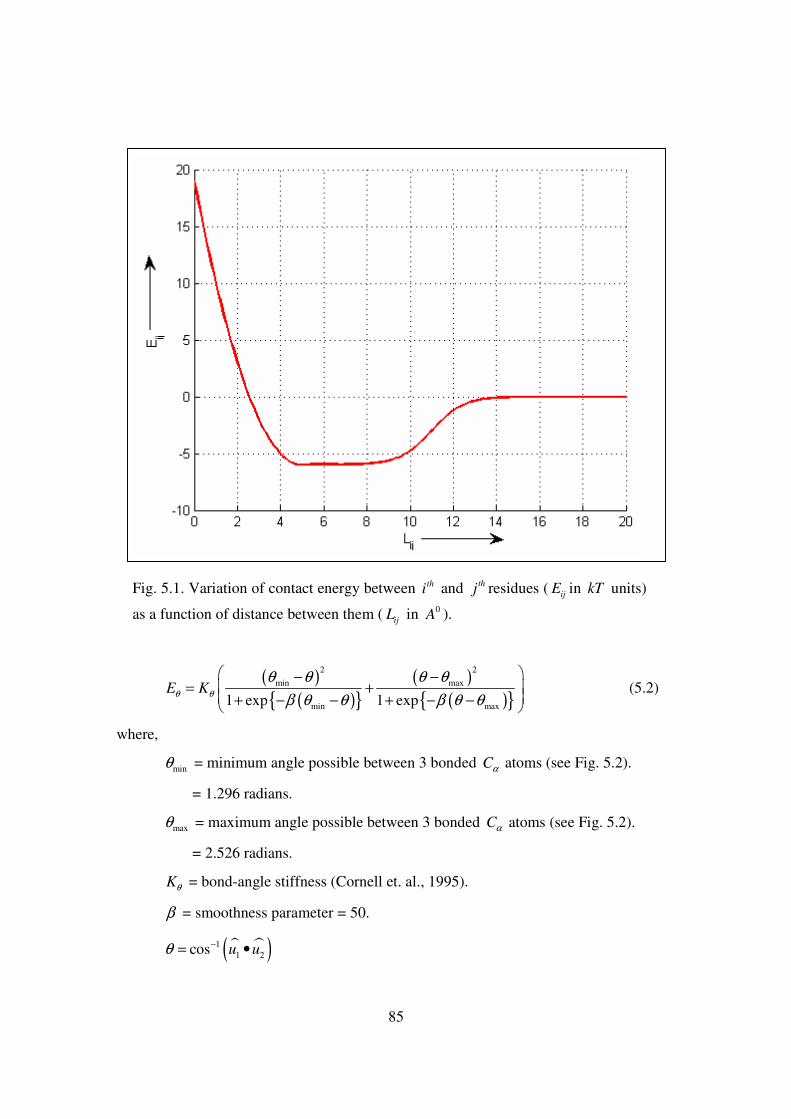
85
( )
( ){ }( )
( ){ }
2 2
min max
min max1 exp 1 expE Kθ θ
θ θ θ θ
β θ θ β θ θ
− −= +
+ − − + − − (5.2)
where,
minθ = minimum angle possible between 3 bonded Cα atoms (see Fig. 5.2).
= 1.296 radians.
maxθ = maximum angle possible between 3 bonded Cα atoms (see Fig. 5.2).
= 2.526 radians.
Kθ = bond-angle stiffness (Cornell et. al., 1995).
β = smoothness parameter = 50.
� �( )11 2cos u uθ −= •
Fig. 5.1. Variation of contact energy between thi and thj residues ( ijE in kT units)
as a function of distance between them ( ijL in 0A ).

86
and �1u and �2u are the unit vectors joining thi and ( 1)th
i − Cα atoms and thi and ( 1)th
i +
Cα atoms respectively. Figure 5.3 shows how the penalty term Eθ for the angle varies as
bond angle θ changes for three bonded Cα atoms. We incorporate another penalty term
to prevent the violation of the fixed bond-length between any two bonded Cα atoms. This
is done using the following function:
( )2
0bonded b ijE K L L= − (5.3)
where,
bK = bond length stiffness (Cornell et. al., 1995).
ijL = distance between th
i and thj bonded Cα atoms.
0L = equilibrium distance two bonded Cα atoms.
= 3.8 Å.
Fig. 5.2. The limits of angle θ between three adjacent Cα atoms. R (magenta
colored dots) represents a residue, CaCa the Cα atom, N (blue colored dots) for
Nitrogen and Coxy the Carboxyl Carbon atom. The Cα atoms on the ends can take
positions within the green cones only. This is due to the 3sp hybrid state of the Cα
atom at the center.
minθ
maxθ
φψ
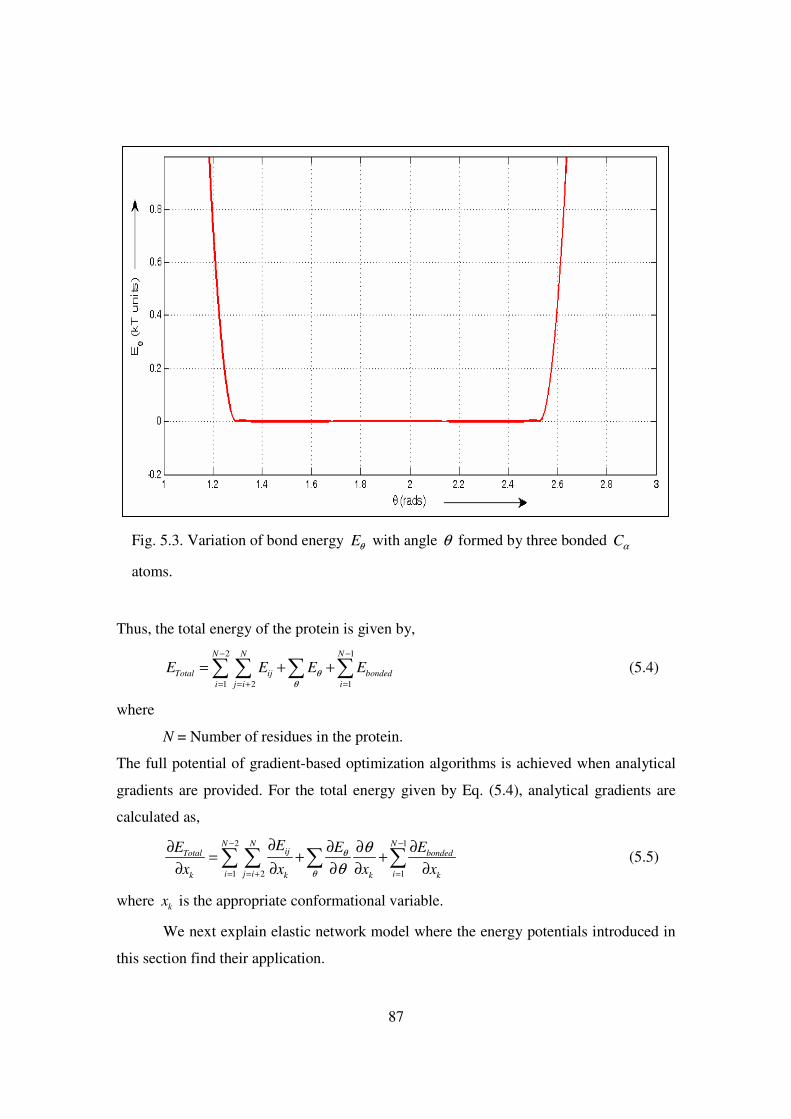
87
Thus, the total energy of the protein is given by,
2 1
1 2 1
N N N
Total ij bonded
i j i i
E E E Eθθ
− −
= = + =
= + +∑ ∑ ∑ ∑ (5.4)
where
N = Number of residues in the protein.
The full potential of gradient-based optimization algorithms is achieved when analytical
gradients are provided. For the total energy given by Eq. (5.4), analytical gradients are
calculated as,
2 1
1 2 1
N N NijTotal bonded
i j i ik k k k
EE E E
x x x x
θ
θ
θ
θ
− −
= = + =
∂∂ ∂ ∂∂= + +
∂ ∂ ∂ ∂ ∂∑ ∑ ∑ ∑ (5.5)
where kx is the appropriate conformational variable.
We next explain elastic network model where the energy potentials introduced in
this section find their application.
Fig. 5.3. Variation of bond energy Eθ with angle θ formed by three bonded Cα
atoms.

88
5.3 Elastic Network (EN) model
5.3.1 Method
Figure 5.4 shows an elastic network (EN) model of a small 10-residue long de novo
protein called Chigolin (PDB ID:. 1UAO). In this model, all the residues are centered at
the Cα atom positions and are connected to one another by imaginary springs. Since the
bond-energies are much higher than the non-bonded interactions, we take the length
between the covalently linked residues to be fixed at 3.8 Å. Hence, the bonded residues
are joined by springs of high stiffness. The non-bonded residues are joined by springs
whose stiffness is given by the absolute value of the MJ contact potential of the
interacting residues. We derive the stiffness matrix K (R D Cook, 2002) of the EN model
in the same manner as a three-dimensional truss structure. Any small deformed shape of
the EN can be expressed as a linear combination of the eigenvectors of K . Thus, if { }0x
is the position vector of all the residues in EN at an initial condition, we can express the
position vectors of all residues { }x of a nearby conformation as,
Fig. 5.4. Elastic network model of a small de-novo protein, Chignolin. The blue
circles represent the amino acid residues centered on their respective Cα atoms.
The covalently bonded residues are connected by black lines. The non bonded
residues are connected by green lines.
θ

89
{ } { } { }3
01
N
i i
i
x x α ω=
= +∑ (5.6)
where,
N = number of residues in the polypeptide.
{ }x = column vector of Cartesian coordinates of all Cα atoms; so its dimension is
3 1N × .
{ } = th
i iω eigenvector of K with dimension 3 1N × .
iα = scalar multiplier associated with thi eigenvector { }iω .
The scalar multipliers { }iα form the set of design variables in our optimization problem
formulation. By varying these coefficients, we change the conformation; and with it the
energy of the polypeptide. This method allows only small changes in conformation
because eigenvector decomposition is restricted to the linear regime. To apply this
method for large changes in conformation, we formulate a novel algorithm which updates
the stiffness matrix of EN of the polypeptide from time to time as optimization
progresses. This algorithm is shown in Fig. 5.5. It is implemented in MATLAB and uses
the optimization toolbox fminunc. As explained in Fig. 5.5, the rate at which we update
the stiffness matrix K is determined by the maximum number of iterations (maxiter)
specified in the optimization program. In Fig. 5.6 we show, for different values of
maxiter, how conformational energy of Chignolin varies with iteration as we minimize
the energy from a fully unfolded state. Table 5.1 compares the energy of the final
conformation, total number of iterations and actual cpu time for different values of
maxiter. It is interesting to note that the energy of the optimal conformation shows
insignificant change as maxiter is varied, as shown in Table 5.1. Even when we do not
update the stiffness matrix at all, the change in optimal value of the energy is very low.
To save time in the case of large polypeptides, one could perform the optimization
without updating K . In such case, one can think of using the eigenvectors as basis

90
Fig. 5.5. Flowchart showing our algorithm for large change in conformation
determined using eigenvectors of stiffness matrix K of EN.
No
Initial conformation: fully stretched polypeptide. Form the stiffness matrix K . Set tolerance =ε . Set initial guess of { }iα s.
Set maximum number of iterations (maxiter) for optimization program fminunc after which K is updated. Calculate energy E . Set E to some very large value.
Run optimization program fminunc. New energy = newE .
Update K with new { }iα s.
Update old energy
newE E= newE E ε− > Yes
Stop iterations. Final energy =
newE .
{ }i newα and eigenvectors of newK define final
conformation.
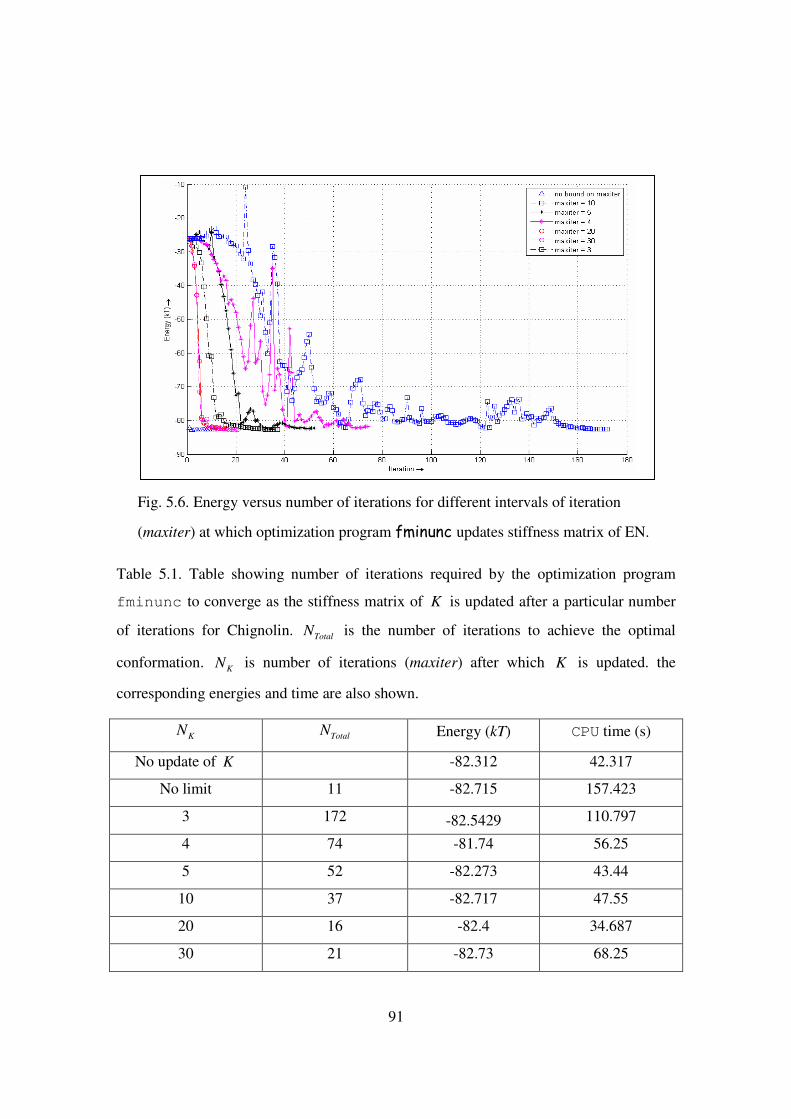
91
Table 5.1. Table showing number of iterations required by the optimization program
fminunc to converge as the stiffness matrix of K is updated after a particular number
of iterations for Chignolin. TotalN is the number of iterations to achieve the optimal
conformation. KN is number of iterations (maxiter) after which K is updated. the
corresponding energies and time are also shown.
KN TotalN Energy (kT) CPU time (s)
No update of K -82.312 42.317
No limit 11 -82.715 157.423
3 172 -82.5429 110.797
4 74 -81.74 56.25
5 52 -82.273 43.44
10 37 -82.717 47.55
20 16 -82.4 34.687
30 21 -82.73 68.25
Fig. 5.6. Energy versus number of iterations for different intervals of iteration
(maxiter) at which optimization program fminunc updates stiffness matrix of EN.

92
vectors for searching the design domain even though their effectiveness is reduced
because of large conformational changes.
5.3.2 Results
We test our formulation for exploring the conformational space and determining local
minima of a few de novo proteins. The first among these is Chignolin (PDB ID: 1UAO).
It is a small 10-residue polypeptide having the amino acid sequence
{G,Y,D,P,E,T,G,T,W,G}. Figure 5.7 shows the conformation of Chignolin in its native
state and after optimizing from the same state. Its native state energy is 77.842 kT− units
(we have done our calculations in kT units as MJ matrix is given in these units;
1 unit = 0.62 /kT kcal mole ) and the radius of gyration is 4.656 Å. The (local) minimum
energy conformation has energy 82.875 kT− and the radius of gyration is 4.139 Å. The
distance root mean square deviation (DRMSD) error (Levitt, 1976) between the two
structures is 1.927 Å. The time taken for optimization on a single processor desktop was
50.515 s. Next, we started with an initial conformation in which Chignolin was fully
unfolded. This is shown in Fig. 5.8a. We also show the native state to bring out the
differences in the two conformations. The unfolded conformation has energy
25.987 kT− and radius of gyration is 10.472 Å. The minimum energy conformation is
shown in Fig. 5.8b. Its energy is 82.717 kT− and the radius of gyration is 4.093 Å. The
Fig. 5.7. Conformation of Chignolin in native state and after optimization. The left
conformation (red lines and blue circles) represent the native state from PDB. The
type of residues are indicated beside respective circles. The right conformation
(blue lines and green circles) represent the optimal one.
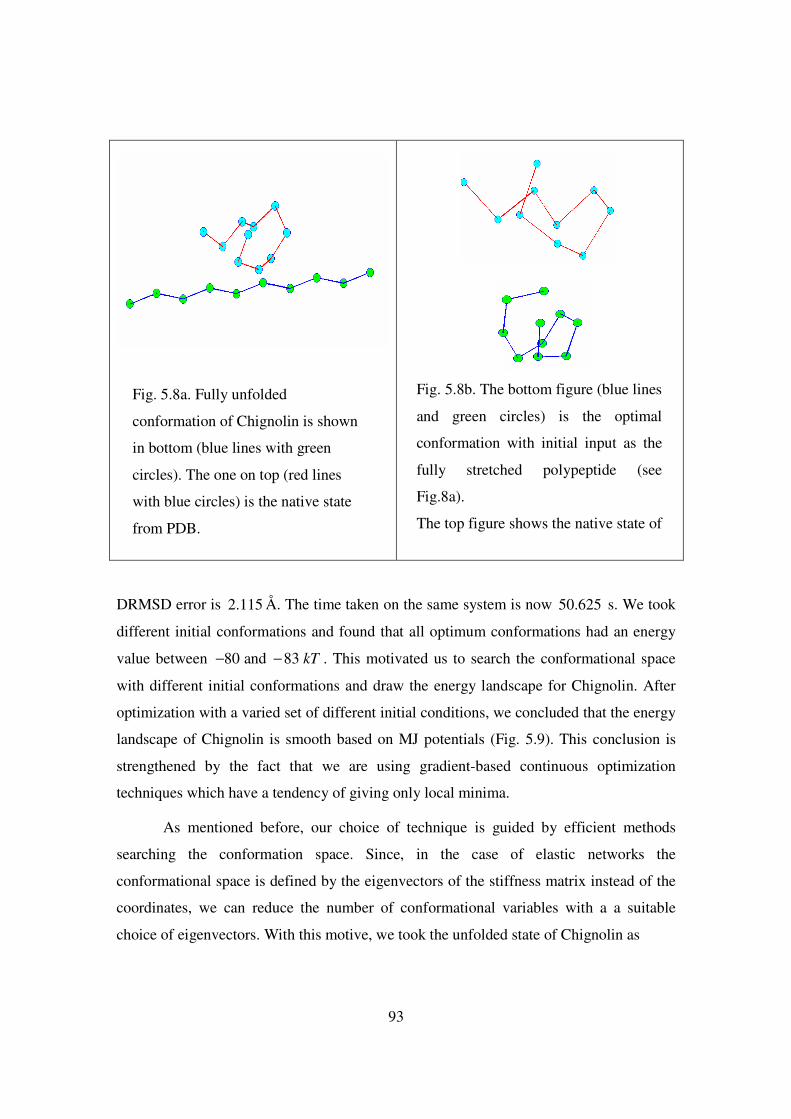
93
DRMSD error is 2.115 Å. The time taken on the same system is now 50.625 s. We took
different initial conformations and found that all optimum conformations had an energy
value between 80 and 83 kT− − . This motivated us to search the conformational space
with different initial conformations and draw the energy landscape for Chignolin. After
optimization with a varied set of different initial conditions, we concluded that the energy
landscape of Chignolin is smooth based on MJ potentials (Fig. 5.9). This conclusion is
strengthened by the fact that we are using gradient-based continuous optimization
techniques which have a tendency of giving only local minima.
As mentioned before, our choice of technique is guided by efficient methods
searching the conformation space. Since, in the case of elastic networks the
conformational space is defined by the eigenvectors of the stiffness matrix instead of the
coordinates, we can reduce the number of conformational variables with a a suitable
choice of eigenvectors. With this motive, we took the unfolded state of Chignolin as
Fig. 5.8b. The bottom figure (blue lines
and green circles) is the optimal
conformation with initial input as the
fully stretched polypeptide (see
Fig.8a).
The top figure shows the native state of
Fig. 5.8a. Fully unfolded
conformation of Chignolin is shown
in bottom (blue lines with green
circles). The one on top (red lines
with blue circles) is the native state
from PDB.
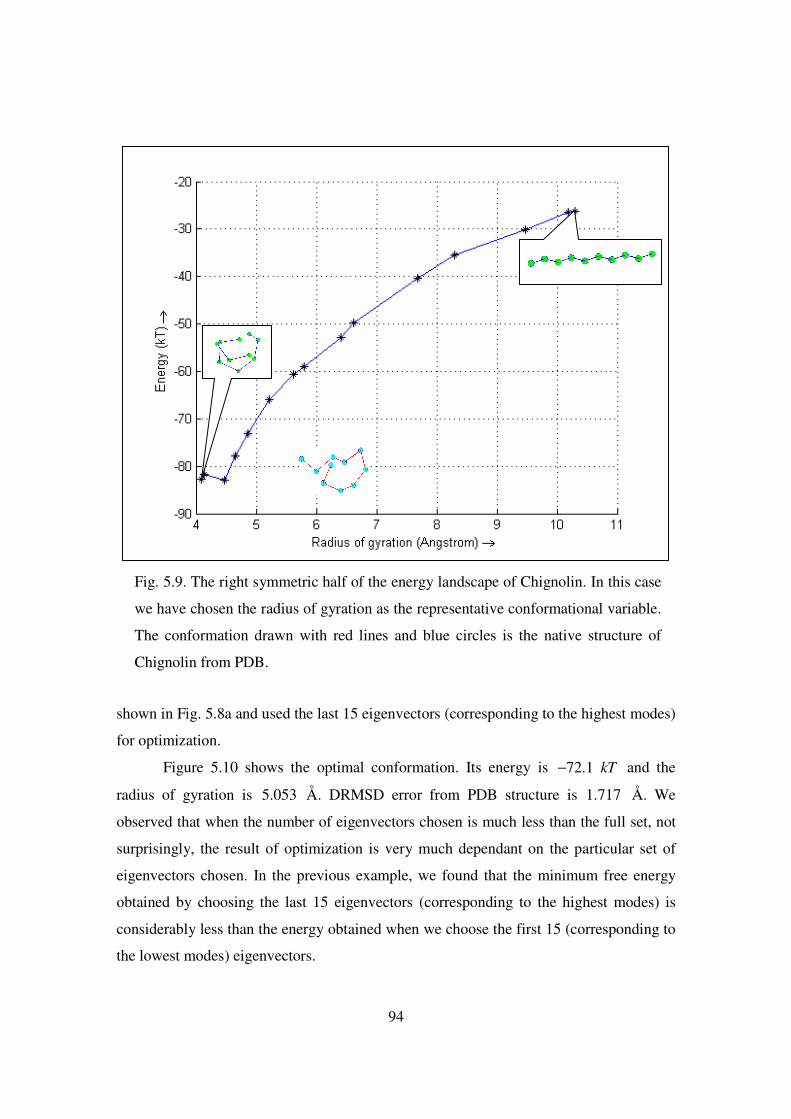
94
shown in Fig. 5.8a and used the last 15 eigenvectors (corresponding to the highest modes)
for optimization.
Figure 5.10 shows the optimal conformation. Its energy is 72.1 kT− and the
radius of gyration is 5.053 Å. DRMSD error from PDB structure is 1.717 Å. We
observed that when the number of eigenvectors chosen is much less than the full set, not
surprisingly, the result of optimization is very much dependant on the particular set of
eigenvectors chosen. In the previous example, we found that the minimum free energy
obtained by choosing the last 15 eigenvectors (corresponding to the highest modes) is
considerably less than the energy obtained when we choose the first 15 (corresponding to
the lowest modes) eigenvectors.
Fig. 5.9. The right symmetric half of the energy landscape of Chignolin. In this case
we have chosen the radius of gyration as the representative conformational variable.
The conformation drawn with red lines and blue circles is the native structure of
Chignolin from PDB.

95
Next, we apply our technique to find the final conformation of a few more de novo
protein sequences. Since Chignolin has a beta hairpin structure, we now select a
polypeptide that contains a helix (PDB ID: 1GJF). This de novo protein contains 14
residues having the sequence {R,A,G,P,L,Q,W,L,A,E,K,Y,Q,G}. The native structure has
energy equal to 152.96 kT− and the radius of gyration is 7.00 Å. We perform
optimization with an initial input as the native structure. The optimal structure has
215.81 kT− energy and the radius of gyration is 4.489 Å. The time taken for
optimization was 71.58 s. When a fully unfolded conformation was taken as the initial
input for optimization the minimum energy was 216.617 kT− . The time taken was
178.013 s. The optimal conformation is shown in Fig. 5.11. The radius of gyration is
4.434 Å. DRMSD from PDB structure is 4.99 Å. We then tried a longer polypeptide
than the two considered thus far. We selected a 23-residue (PDB iID: 1RIJ) as our next
target. 1RIJ has energy 405.2334 kT− in native state (PDB conformation) and radius of
gyration is 7.151 Å. The optimal conformation for 1RIJ starting form fully unfolded
state is shown in Fig. 5.12. It’s energy is 580.146 kT− and the radius of gyration is
5.048 Å. The time taken was 24 min and 31.1 s. DRMSD from PDB conformation is
4.966 Å.
Fig. 5.10 b. Optimal conformation
using last 15 eigenvectors of EN
matrix.
Fig. 5.10 a. The native state of
Chignolin (PDB ID 1UAO).

96
Fig. 5.11a. PDB structure of 1GJF. Fig. 5.11b. Optimal structure of
1GJF from fully unfolded state.
Fig. 5.12 a. Native structure (PDB)
of 1RIJ.
Fig. 5.12 b. Optimal structure of
1RIJ from fully unfolded state.

97
We now take two natural proteins, namely, Ubiquitin (PDB ID: 1UBQ) with 76 residues,
and Lysozyme with 164 residues. Since, there was not much difference in results even
when K was not updated periodically, for these two cases we applied optimization
without updating K in order to save time. As discussed earlier, we can take the
eigenvectors of K as the basis vectors of the conformation space. When we perform
optimization in this space, there was no need to change the basis vectors of the space. The
optimal conformation and the native state of Ubiquitin from PDB are shown in Fig. 5.13.
Time taken for optimization was 23 h 29 m and 32 s. The energy of native conformation
was 2245.3 kT− with the radius of gyration equal to 11.493 Å. The energy after
optimization is 6830.5 kT− and radius of gyration is 5.576 Å. RMSD error from PDB
structure is 9.142 Å. The PDB conformation of Ubiquitin is far away from what we got
as the local minimum-energy conformation. We observe that our conformation is much
more compact than the PDB structure. This is because in the optimal conformation we do
not have secondary structures such as alpha helices and beta sheets. Secondary structures
Fig. 5.13 a. PDB structure of
Ubiquitin; the secondary structures
have been shown with bold lines.
Fig. 5.13 b. Conformation after
minimization of energy from native
state.

98
are integral components of protein conformation. However, our energy model does not
contain any function to simulate the formation of hydrogen bonds which are crucial for
the formation of secondary structures.
For predicting the structure of Lysozyme, we start with an unfolded state keeping
the secondary structures intact. The DRMSD of the unfolded state from which we start
our simulation from the native state is 19.643 Å and the radius of gyration is 27.815 Å.
The radius of gyration of the native state of Lysozyme is 16.244 Å. We did two sets of
simulations for Lysozyme, once by taking the full set of eigenvectors, which is 216, and
then by taking a reduced set of 130 eigenvectors. When we take the full set of
eigenvectors and start the optimization from an unfolded state, the optimal conformation
has DRMSD of 12.43 Å from the native state and its radius of gyration is 21.234 Å. The
time required for this simulation on a Xeon 3.0 GHz desktop computer is almost a week.
With the reduced set of 130 eigenvectors, we did two simulations, once by taking the first
130 and then by taking the last 130 eigenvectors. When we take the first 130
eigenvectors, the optimal conformation has DRMSD of 13.427 Å from PDB and its
radius of gyration is 22.548 Å. When we take the last 130 eigenvectors, the optimal
conformation has DRMSD of 17.447 Å from PDB and its radius of gyration is 25.704 Å.
In this case, the energy obtained with first 130 eigenvectors is much less than that
obtained with last 130 eigenvectors. This is in complete contrast with the results on
Chignolin, where the last set of eigenvectors chosen gave less energy than the first set of
eigenvectors. Thus, we cannot comment on predicting which set of eigenvectors will give
an optimal conformation with lower energy. The time required for simulation was 2 days
9 h and 10 min on a Xeon 3.0 GHz desktop computer.
In the next section, we give a brief description about the formulation of a
continuous function which can predict OB-CG model of helices starting from the fully
unfolded state.
5.3 Secondary structure formation using continuous optimization
As mentioned in the introduction of this thesis, secondary structures in protein molecules,
such as alpha helices and beta sheets, form the key structural constituents of the structure
of proteins. Without secondary structures, a polypeptide will not be classified as a
protein. Hence, we tried to incorporate a function which would enable the formation of

99
secondary structures based on our coarse-grained model. The following is a continuous
function which when optimized with respect to the conformation variables (the
coordinates of the residues) forms a helix from a fully unfolded polypeptide.
( ){ } ( )4
22 cos( ), 4
1 1
h
k
N N
HB HB k HB i i p
k i
E e e e L Lθ
θθ ω−
−
+= =
= − + + −∑ ∑ (5.7)
where,
HBe = strength of hydrogen bond in alpha helices (Arora and Jayram, 1997).
kθ = the kth dihedral angle between the planes formed by Cα atoms i, i+1, i+2 and
i+1, i+2, i+3 (Fig. 5.14).
ω = ideal coarse grained dihedral angle to form alpha helices (Tozzini and
Rocchia, 2006).
Fig. 5.14. Schematic diagram of an alpha helix; the Cα atoms are shown as black
dots. The bonds facing the viewer are drawn in thick black lines and the ones away
from the viewer are drawn dotted. �1v is the unit vector normal to the plane formed
by the i, i+1 and i+2 Cα atoms. �2v is the unit vector normal to the plane formed by
the i+1, i+2 and i+3 Cα atoms.
i +2
i
i +1
i +3
�1v
�2v
i +4

100
Nθ = number of coarse grained consecutive dihedral angles in the helix.
hN = number of residues in the helix.
, 4i iL + = distance between the ith and (i+4)th Cα atoms (Fig. 5.14).
pL = pitch distance between two Cα atoms in the coarse grained model (Tozzini
and Rocchia, 2006).
When we optimize HBE in Eq. 5.7 with bond angle penalty (Eq. 5.2) and bond length
penalty (Eq. 5.3) we get helices (Fig. 5.15b) from the fully unfolded state (Fig. 5.15a).
However, when we add the non-bonded interaction energy term (Eq. 5.1) from our
coarse-grained energy model, the helical structures are no longer formed after
optimization, as shown in Fig. 5.15c.
For beta sheets, we not only have to formulate a function to form beta strands, but
also another function which will pair the strands. The problem of pairing of β -strands to
form β -sheets is combinatorial in nature, and has been termed as a “particularly difficult
task” (Yue and Dill, 2008). In this thesis, it has not been possible to give a continuous
function which simultaneously forms beta strands and also aids in their pairing.
Researchers have used other techniques, for example, global optimization (Klepeis and
Floudas, 2003) and machine learning (Aydin et al., 2011) to predict the pairing of β -
strands to form β -sheets. We would like to mention here that the formulation for β -
strands to form β -sheets using continuous functions that are amenable to gradient based
optimization techniques is still open. Secondary structure prediction form sequence
(alpha helix and beta strands) has been solved using various machine learning techniques
and to date a number of web-based servers are available which predict secondary
structures from a sequence (www.expasy.ch/tools/#secondary). Since our simultaneous
sequence and conformation search requires prediction of native-like folded tertiary
structures from the sequence, henceforth we consider the secondary structures as rigid
bodies which can be predicted using web-based servers from the sequence in the
conformation search program.
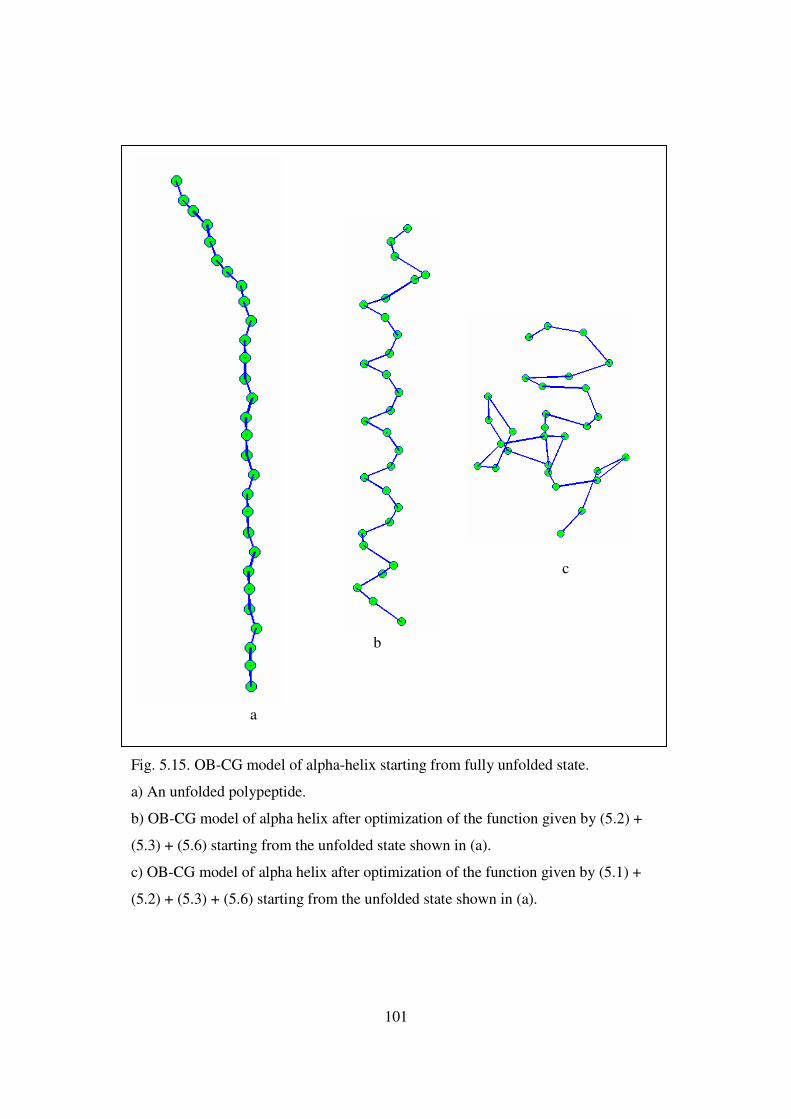
101
b
a
c
Fig. 5.15. OB-CG model of alpha-helix starting from fully unfolded state.
a) An unfolded polypeptide.
b) OB-CG model of alpha helix after optimization of the function given by (5.2) +
(5.3) + (5.6) starting from the unfolded state shown in (a).
c) OB-CG model of alpha helix after optimization of the function given by (5.1) +
(5.2) + (5.3) + (5.6) starting from the unfolded state shown in (a).

102
5.4 Conformation search using coarse-grained model with rigid secondary
structures
5.4.1 Method
In this section, we model protein folding as the folding of a three-dimensional linkages or
a chain under the action of coarse-grained molecular forces (Fig. 5.16). As mentioned in
the last section, we consider the secondary structures such as alpha helices as rigid bodies
in our model. This assumption is supported by the framework model of protein folding
which hypothesizes that the tertiary structure is formed by the packing of secondary
structures that are formed during the initial stages of folding (Karplus and Weaver, 1994,
Fain and Levitt, 2003, Gong and Rose, 2005, Fleming et al., 2006, Rose et al., 2006, Wu
et al., 2008). Since the secondary structures are formed before folding, they can be
considered as rigid bodies in the simulation (Erman et al., 1997, Yue and Dill, 2008,
Nanias et al., 2003, Sancho et al., 2004). However, in this work we present our result on
proteins consisting of only α -helices in which we consider the α -helices as rigid bodies.
We use the nonlinear conjugate gradient algorithm (refer Appendix B3) to
minimize the energy potentials and determine the minimum energy conformations. The
conjugate gradient algorithm can solve a convex problem of n variables in O(n) steps. We
code the algorithms in C++ language and use a 3.0 GHz Xeon quad-core computer to
implement our programs. We predict the structures of seven proteins of varying chain
lengths (from 36 to 164 residues) using the two energy models mentioned before (the
OB-CG model described in section 5.2 and Levitt’s OB-CG model).
Here we briefly describe the terms that we have used from Levitt’s coarse-grained
model (Levitt, 1976). In Levitt’s model (Levitt, 1976) the non-bonded interaction energy
is given by,
8 60 0
3 4ij ij
ij ij
ij ij
r rE
r rε
= −
(5.8)
where,
ijr = distance between thi and th
j residues = ( ) ( ) ( )2 2 2
i j i j i jx x y y z z− + − + −
ijε and 0ijr are parameters which depend on the type of residues at positions i and j.
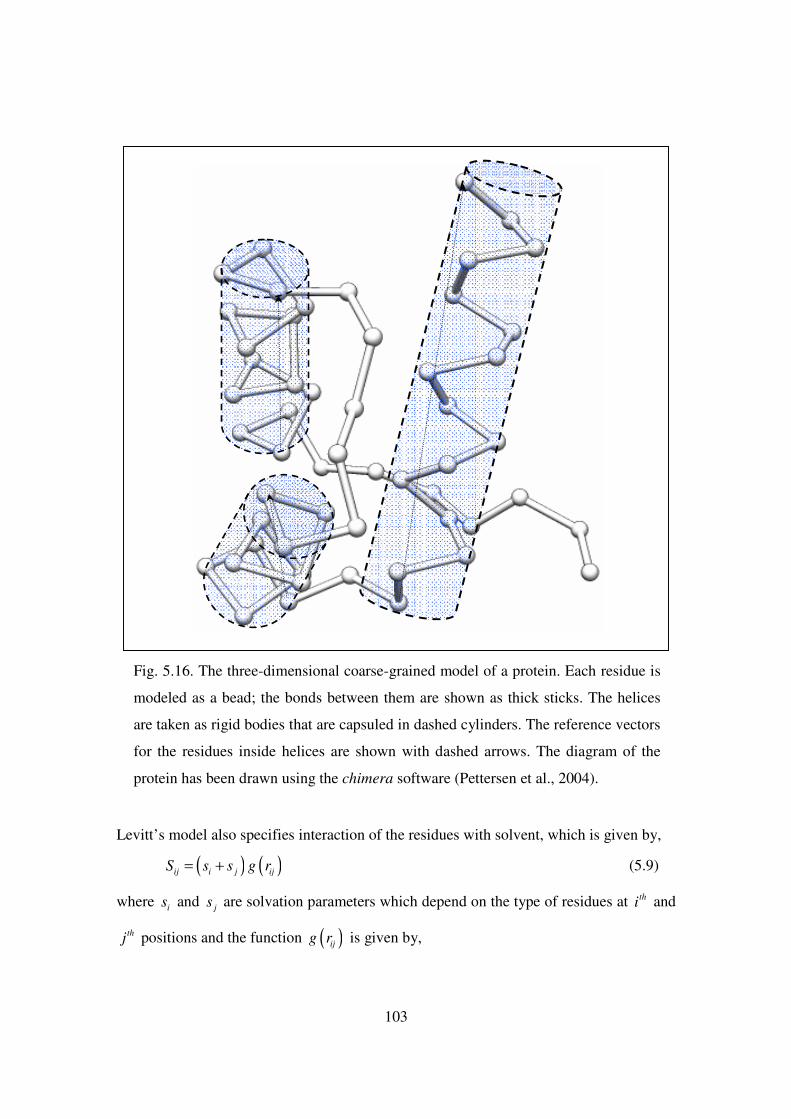
103
Levitt’s model also specifies interaction of the residues with solvent, which is given by,
( ) ( )ij i j ijS s s g r= + (5.9)
where is and
js are solvation parameters which depend on the type of residues at thi and
thj positions and the function ( )ijg r is given by,
Fig. 5.16. The three-dimensional coarse-grained model of a protein. Each residue is
modeled as a bead; the bonds between them are shown as thick sticks. The helices
are taken as rigid bodies that are capsuled in dashed cylinders. The reference vectors
for the residues inside helices are shown with dashed arrows. The diagram of the
protein has been drawn using the chimera software (Pettersen et al., 2004).

104
( ) ( )2 4 6 811 7 9 5
2ijg r x x x x= − − + − for 1x < (5.10)
0= for 1x ≥
where
9.0
ijrx =
Thus, the non-bonded energy using Levitt’s potential is given by ij ijE S+ . We have
omitted other terms in Levitt’s potential as those are not amenable to our OB-CG model.
5.4.2 Results
We take a set of seven proteins of different chain lengths to test the efficacy of the
gradient-based optimization method in predicting possible native-like protein structures.
The PDB IDs of each protein with the corresponding number of residues are given in
Table 5.2. Since, the optimization function (Eq. 5.4) is nonconvex, there ought to be
multiple minima. However, gradient-based optimization methods, even though very
efficient in determining local minima, cannot escape from a local minimum. Hence, we
search for minimum conformations using different initial structures (inputs to
optimization), i.e., structures which obey the constraints of the protein backbone (bond
length and angle constraints) but are conformationally well stretched as compared to the
native state, which is compact. We used 50-60 initial structures for proteins with residues
less than 50, 30-40 initial structures for proteins with residues between 50 and 100, and
20-25 initial structures for roteins with residues greater than 100.
Figure 5.17 shows the predicted structure of a protein with 81 residues (PDB ID:
1LRE). On the left hand side of the figure we show the predicted backbones using our
OB-CG potential (Fig. 5.17b) and Levitt (Fig. 5.17c) as also the native structure from
PDB (Fig. 5.17a). Since we have considered the α -helices as rigid bodies in our model,
the same are indicated with a different color (blue) on the backbone of the protein. As it
is difficult to understand the similarities/differences between three-dimensional chains of
proteins on a two-dimensional plane, we also show the contact map for each structure on
the right hand side. On the contact map, we consider the residue numbers along the X and
105
a
c
b

106
Y axes. If two non-bonded residues are within a particular distance (the contact distance,
which we take as 6.5 Å based on the MJ matrix) from each other a dot is placed on the
map at the corresponding position. For example, if residues i and j are within contact
distance, then we place a dot at the positions (i,j) and (j,i) on the map. The contact map is
very helpful in noting non-bonded residues which are far apart along the length of the
chain, but are in close proximity spatially. Such contacts indicate favorable interaction
among residues and are crucial in determining the tertiary or quaternary structure of the
protein. It is interesting to note that the α -helices appear as bands placed along the
diagonal in the contact maps (Figs. 5.17a, 5.17b, and 5.17c).
Table 5.2 shows the results of predicted structures for all the proteins. The first
column indicates the PDB ID, the number of residues in the protein, and also the
percentage of residues that are in the secondary structures, namely the α -helices. We do
this so that the reader can appreciate the simplification in computation once the α -
helices are assumed as rigid bodies in the simulation. In Table 5.2, we show the
DRMSDs for the predicted structure of each protein from corresponding native structures
Fig. 5.17. Predicted and native state structures of the protein with PDB id. 1LRE (81
residues). On the left hand side, we show the coarse-grained representation of the
protein (made with chimera), in which each residue is shown as a bead on the
backbone. The alpha helices are colored blue. On the right hand side we show the
corresponding contact map for the structure. Two non-bonded residues are taken to
be in contact if they are within a distance of 6.5 Å from each other. The contact
maps have been drawn using MATLAB software.
a) Native structure of the protein with PDB ID 1LRE. The contact map is shown on
the right hand side with black colored dots.
b) Minimum energy structure of the same protein using our OB-CG function. The
contact map is shown on the right hand side with red colored dots.
c) Minimum energy structure of the same protein using Levitt’s potential. The
contact map is shown on the right hand side with blue colored dots.
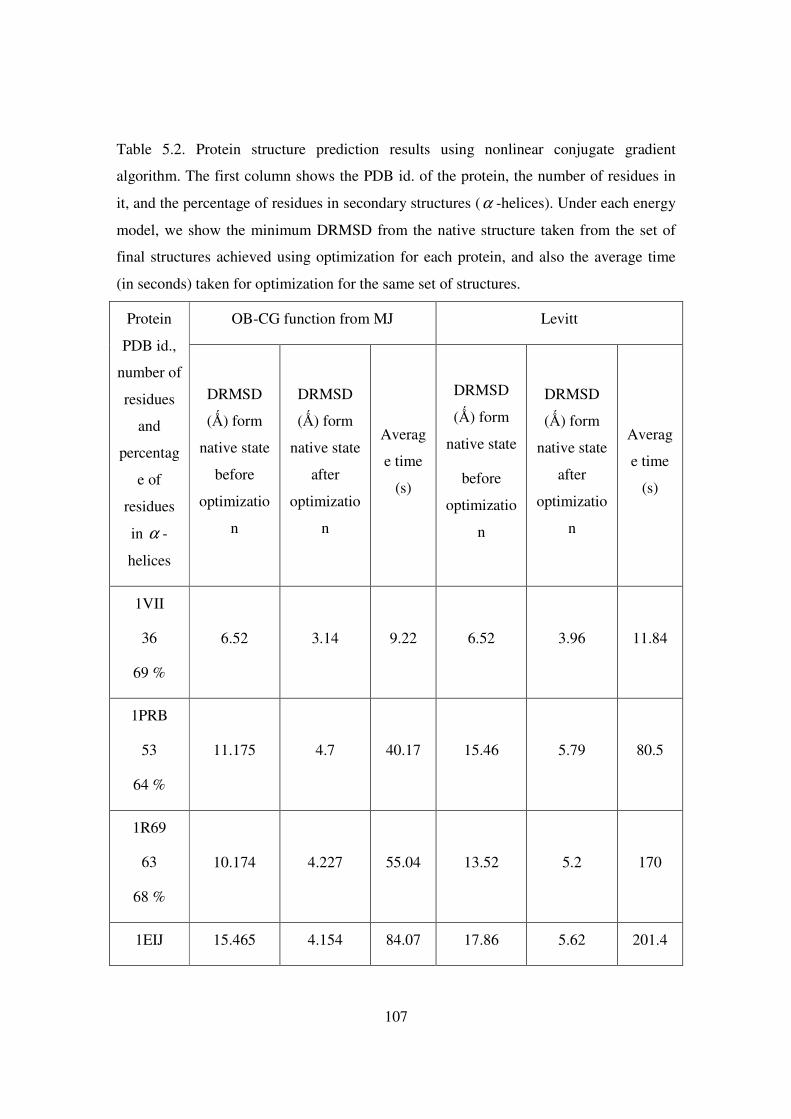
107
Table 5.2. Protein structure prediction results using nonlinear conjugate gradient
algorithm. The first column shows the PDB id. of the protein, the number of residues in
it, and the percentage of residues in secondary structures (α -helices). Under each energy
model, we show the minimum DRMSD from the native structure taken from the set of
final structures achieved using optimization for each protein, and also the average time
(in seconds) taken for optimization for the same set of structures.
OB-CG function from MJ Levitt Protein
PDB id.,
number of
residues
and
percentag
e of
residues
in α -
helices
DRMSD
(Ǻ) form
native state
before
optimizatio
n
DRMSD
(Ǻ) form
native state
after
optimizatio
n
Averag
e time
(s)
DRMSD
(Ǻ) form
native state
before
optimizatio
n
DRMSD
(Ǻ) form
native state
after
optimizatio
n
Averag
e time
(s)
1VII
36
69 %
6.52 3.14 9.22 6.52 3.96 11.84
1PRB
53
64 %
11.175 4.7 40.17 15.46 5.79 80.5
1R69
63
68 %
10.174 4.227 55.04 13.52 5.2 170
1EIJ 15.465 4.154 84.07 17.86 5.62 201.4

108
72
68 %
1LRE
81
74 %
14.79 3.886 68.17 17.4 6.05 237.8
1BCF
(chain A)
158
78 %
55.13 6.66 810.08 22.7 8.9 3599.6
1LYD
164
65 %
29.8 8.65 288.24 22.9 9.9 1926.6
using both energy models. Here, we only show the minimum DRMSDs that we get for
each protein from the set of different initial structures. To convey to the reader a sense of
the unfoldedness of the structures that we use as initial inputs (which are not shown
using three-dimensional figures or contact maps), we also show the DRMSD of the
corresponding initial structures for all proteins for both energy models. One can note
from Table 5.2 that gradient-based optimization methods are able to achieve a two to
four-fold reduction in DRMSD from the native structure. We also show the average time
(in seconds) taken for optimization of each protein structure for both the energy models
using the nonlinear conjugate algorithm.

109
We now take up a few examples in which we demonstrate how this method can be used
for ab initio structure prediction as well. We select three α -helical proteins from our set
and use the hierarchic neural networks (HNN) online server (Guermeur, http://npsa-
pbil.ibcp.fr/cgi-bin/npsa_automat.pl?page=npsa_nn.html) for secondary structure
prediction (to see a short discussion on secondary structure prediction servers refer to
Appendix B4 ). The result of the secondary structure predictions using the HNN server is
shown in Fig. 5.18. We next use Tozzini’s parameters (Tozzini and Rocchia, 2006) to
construct OB-CG secondary structural models (α -helices). These secondary structures
are then connected in order by Cα atoms to give a fully unfolded coarse-grained structure
of the protein. We generate an ensemble of unfolded structures by perturbing the fully
unfolded structures and then apply our optimization program on these structures. The
minimum DRMSD of the final structures from native structure for each protein is given
in Table 5.3.
Fig. 5.18. Predicted and native secondary structures for the proteins 1BCF (chain A),
1EIJ, 1LYD and 1R69. The secondary structures are predicted using the HNN server h –
helix; e – beta strand; c – coil;
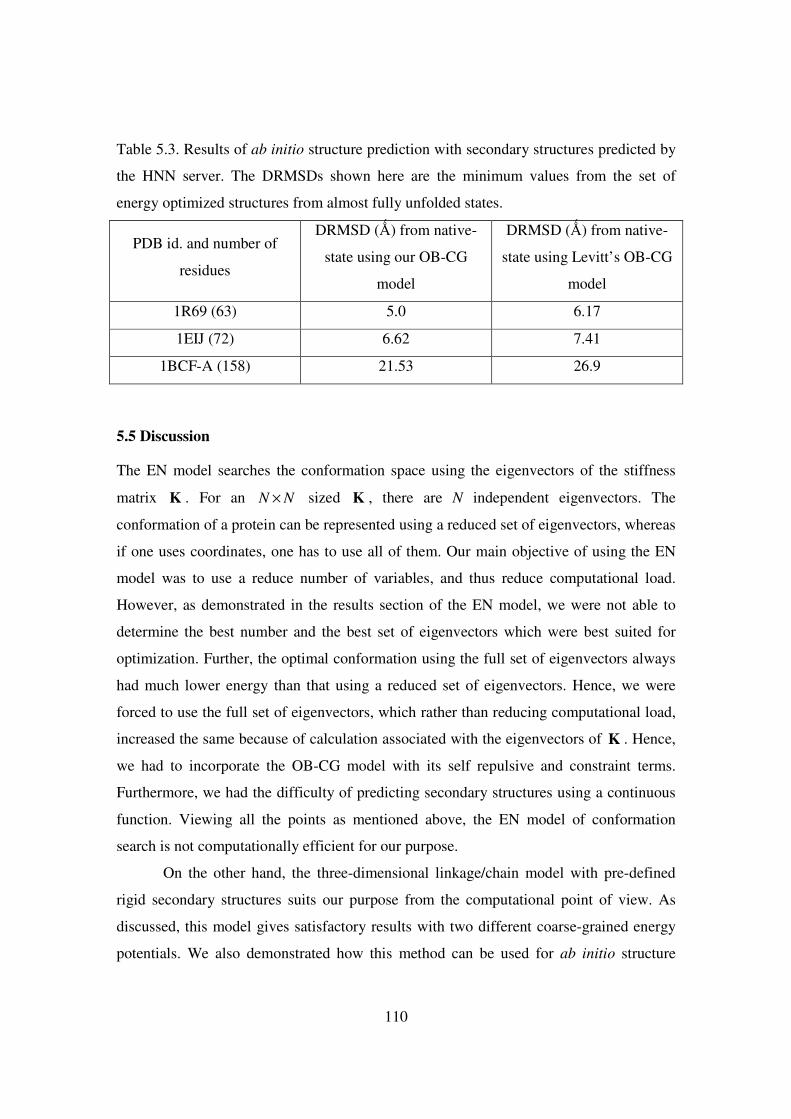
110
Table 5.3. Results of ab initio structure prediction with secondary structures predicted by
the HNN server. The DRMSDs shown here are the minimum values from the set of
energy optimized structures from almost fully unfolded states.
PDB id. and number of
residues
DRMSD (Ǻ) from native-
state using our OB-CG
model
DRMSD (Ǻ) from native-
state using Levitt’s OB-CG
model
1R69 (63) 5.0 6.17
1EIJ (72) 6.62 7.41
1BCF-A (158) 21.53 26.9
5.5 Discussion
The EN model searches the conformation space using the eigenvectors of the stiffness
matrix K . For an N N× sized K , there are N independent eigenvectors. The
conformation of a protein can be represented using a reduced set of eigenvectors, whereas
if one uses coordinates, one has to use all of them. Our main objective of using the EN
model was to use a reduce number of variables, and thus reduce computational load.
However, as demonstrated in the results section of the EN model, we were not able to
determine the best number and the best set of eigenvectors which were best suited for
optimization. Further, the optimal conformation using the full set of eigenvectors always
had much lower energy than that using a reduced set of eigenvectors. Hence, we were
forced to use the full set of eigenvectors, which rather than reducing computational load,
increased the same because of calculation associated with the eigenvectors of K . Hence,
we had to incorporate the OB-CG model with its self repulsive and constraint terms.
Furthermore, we had the difficulty of predicting secondary structures using a continuous
function. Viewing all the points as mentioned above, the EN model of conformation
search is not computationally efficient for our purpose.
On the other hand, the three-dimensional linkage/chain model with pre-defined
rigid secondary structures suits our purpose from the computational point of view. As
discussed, this model gives satisfactory results with two different coarse-grained energy
potentials. We also demonstrated how this method can be used for ab initio structure

111
prediction starting from the sequence with the aid of secondary structure prediction
servers. Henceforth, in our simultaneous sequence and conformation search problem, we
use this strategy of structure prediction from a sequence.
5.6 Closure
In this chapter, we described the formulation of our coarse-grained energy potential based
on the MJ matrix and two methods of searching the conformation space. We also
described the formulation of a continuous function, which when optimized, gives coarse-
grained model of alpha helices starting from a fully unfolded state of polypeptides. Both
the conformation search methods are validated for structure prediction by considering
proteins of different chain sizes. Based on the performance of these methods, we selected
a method for using in the simultaneous search of the sequence and conformation spaces.

112
6. Simultaneous search in the sequence and
conformation spaces: an application
• We consider the re-design of an existing enzyme as a demonstration of our
simultaneous sequence and conformation search strategy.
• A brief description of the target protein, the hen egg-white lysozyme, is given.
• We describe the modeling of the target protein and present the results.
• We discuss the computational methods and the simultaneous sequence and
conformation search strategy in the light of the results.
• The chapter is concluded with a summary.
6.1 Introduction
In this chapter, we present an application of the sequence and conformation search
techniques that are presented in the last two chapters. We combine these methods to
develop a step-by-step procedure for simultaneous sequence and conformation search for
computational protein design as mentioned in the problem statement section of the first
chapter. We take up the computational re-design of an actual protein, the hen-egg white
lysozyme (PDB ID 1LZE). The re-design of an existing protein was taken up because of
lack of access to experimental knowledge and facilities to design an actual protein that
can be experimentally validated. As mentioned in the introduction chapter, the goal of
our work is to give a method of protein design with structural and functional
specifications using gradient-based continuous optimization techniques. The structural
and functional specifications can be posed as constraints in the optimization problem.
Thus, if a few amino acids are crucial for a specific function of the target protein, those
type of amino acids are fixed in the corresponding positions in the sequence while we
search the sequence space for minimum-energy sequences. Again, if a part of the
conformation is important for the function of the target protein, for example, a ligand or
substrate-binding site, that part of the protein conformation can be modeled as a rigid
structure while we search the conformation space for minimum-energy conformations.
The aim of combining the sequence and conformation search techniques is that when
used iteratively, we should achieve convergence both in sequence and conformation

113
spaces if the target protein were to exist, provided all the energy potentials that we use in
our calculations can capture the protein folding phenomenon. The problem of re-design
of an existing protein thus serves as a validation test for the strategy that we develop in
the first chapter.
Enzymes are an important class of proteins that catalyze many biological
reactions. The catalysis takes place by the binding of the reactive molecules (substrates)
to specific pockets on the enzyme, known as the active site of the enzyme (see Fig. 6.1).
Within the active site, a few amino acids form bonds with the substrate resulting in an
enzyme-substrate complex. The enzyme-substrate complex lowers the energy barrier and
thus changes the rate of the reaction in the range of 5 to 17 orders of magnitude (Nelson
and Cox, 2008). The active site and the residues that form bonds with the substrate to
form the enzyme-substrate complex constitute an ideal example of structural and
functional specifications, which may be posed for functionalistic de novo protein design
Active site
Key residues that
take part in reaction
forming the
enzyme-substrate
complex
Fig. 6.1 Schematic of an enzyme molecule. The active site is shown with bold-
dashed lines. Two key residues that form the enzyme substrate complex are shown
with red and blue colored beads.

114
problem. This motivates us to consider the re-design of an enzyme, namely, the hen egg-
white lysozyme (PDB ID 1LZE), as an application of our computational protein design
strategy.
Before embarking on the application of our simultaneous sequence and
conformation search method and presentation of the results that we have achieved, we
would like to convey to the reader the broad perspective with respect to which this work
should be judged. As mentioned in the introductory chapter, developing computational
methods for functionalistic de novo protein design is a challenging problem (Baker,
2010). The difficulty of the problem can be understood when one realizes that both ab
initio structure prediction and de novo sequence design, the key constituents of
functionalistic de novo protein design (Schueler-Furman, 2005), are open problems by
themselves, and any generalized computational method for both these problems has not
yet been developed. Furthermore, each technique, when combined with the other, result
in issues for which the corresponding techniques were not developed individually. For
example, generating sequences and rotamer libraries for flexible backbone templates, and
ab initio structure prediction for large libraries of designed sequences, may encounter
problems when they are used in conjunction with other techniques. The best performing
methods for one problem (e.g., de novo sequence design) when used to address the larger
problem (i.e., simultaneous search of sequence and conformation spaces) results in a
computational deadlock. For instance, using the DEE method for de novo sequence
design for flexible backbone templates is computationally intractable. Thus, one of the
ways of approaching the larger problem will be to use models and techniques which may
give less accurate results but are computationally affordable. The work done in this thesis
takes this approach.
We have developed computational models and methods which are
computationally efficient and can be implemented on desktop computers, but, the
computational efficiency comes at the cost of accuracy. Each of these methods, i.e.,
sequence and conformation search techniques, were tested individually by considering
several examples. Now, we combine these methods; the results are a combination of the
pros and cons inherent in the methods and models that we use. We would further like to
point to the reader that the aim of this thesis is not the computational re-design of the hen

115
egg-white lysozyme (PDB ID 1LZE). The hen egg-white lysozyme is a good textbook
example of an enzyme whose activity and structural details are well understood (Nelson
and Cox, 2008). Furthermore, because of certain structural details which appear to be
conducive to our method (we discuss these details in the relevant section on modeling),
we consider it as an example of the application of the sequence and conformation search
techniques that we have developed. However, there are certain structural details of 1LZE
that have not been accounted for in our model; for example, pairing of Cystine residues to
form disulphide bonds. Thus, this example of application of our technique should be
judged more from the generalized computational point of view and less from the
convergence of results towards the particular protein taken up as a demonstrative
example.
6.2 A brief description of the target protein: The hen egg-white Lysozyme
The hen-egg white lysozyme (PDB ID 1LZE) is a 129 residue enzyme (see Fig. 6.2a) that
cleaves the carbohydrate peptidoglycan found in the cell walls of many bacteria (Nelson
and Cox, 2008). This protein was the first among enzymes to have its three-dimensional
structure determined by David Phillips and colleagues in 1965. There are four disulphide
bonds between Cystine pairs at the following positions: 6-127, 30-115, 64-80 and 76-94
(Maenaka et al., 1995). The key catalytic amino acid residues are Glu35 and Asp52 which
form intermediate bonds with the C—O bond between the N-acetylmuramic (Mur2Ac,
also referred as NAM) and the N-acetylglucosamine (GlcNAc, also referred as NAG)
sugar residues in the peptidoglycan molecule, ultimately leading to the cleavage of the
abovementioned C—O bond. Two different reaction pathways elucidating the enzymatic
action of this lysozyme have been proposed, the SN1 (Phillips mechanism) and the SN2
pathways (Withers et al., 2001). The details of these pathways can be found in relevant
texts (Nelson and Cox, 2008). At present, the SN2 pathway is more in agreement with
experimental results (Nelson and Cox, 2008). The sequence and the secondary structures
of 1LZE are shown in Fig. 6.2b.

116
Fig. 6.2a. A ribbon diagram of the hen egg-white lysozyme (PDB ID 1LZE). The
key catalytic residues, namely, Glu35 and Asp52 are shown as ball and stick models.
The portion of the structure which is assumed to have a fixed conformation is
colored blue.
b. The wild-type sequence of the hen egg-white lysozyme (PDB ID 1LZE). The
secondary structures are shown below the sequence: h – Alpha helix; e – Beta
strand.
Asp52
Glu35
(a)
(b)

117
6.3 Modeling and results
We follow the step-by-step design procedure for computational re-design of 1LZE as
outlined in Fig. 1.7 and in the problem statement section of the introduction chapter. The
problem statement requires that a few residues and a part of the conformation be
specified as inputs to the problem. In the case of 1LZE, we take Glu35 and Asp52 as the
specified residues which are fixed at 35th and 52nd positions respectively in the sequence
while designing the same. For the other specified quantity, i.e., a part of the
conformation, we chose the part of the conformation occupied by residues numbering
from 42 to 60. This is that part of the conformation where a β -sheet is formed from
pairing of three β -strands (shown in blue color in Fig. 6.2a). Although this part of the
conformation is spatially situated close to the active site and may play a role in the
stabilization of the same, we admit that we chose this part because our tertiary structure
prediction program cannot predict pairing of β -strands to form β -sheet. Thus, the
choice of the part of the conformation occupied by residue numbers 42-60 was more
guided by computational considerations than by biological significance. Here, we would
also like to point out that one of the reasons of selecting 1LZE as the target protein is
because of dominance of α -helices among its secondary structures (Fig. 6.2 b).
At present, our design method can consider only single-chain proteins. Thus,
before starting design, we consider the single chain information of 1LZE as a given
information. Furthermore, since this design strategy works for a fixed number of design
variables, the number of residues in the protein, i.e., 129, is also assumed to be a
specified quantity. Hence, the following are the inputs to the re-design of 1LZE:
i. the number of chains: 1.
ii. the number of residues: 129.
iii. fixed residues: Glutamic acid and Aspartic acid at positions 35 and 52
respectively.
iv. fixed conformation: The Cα coordinates from residue numbers 42-60.
v. apart from the fixed conformation the target protein comprises predominantly α -
helices, and

118
vi. the amino acid composition, i.e., number of amino acids of each type, are same as
in the wild-type sequence.
With these inputs, the first step is to design sequences based on free energy minimization.
However, at this point we know only the conformation of the backbone for residue
numbers 42-60. In this stage (step (i) in Fig. 1.7) we use the double sigmoid method with
Zhang and Skolnick’s potentials (Zhang and Skolnick, 1998) to design sequences. Since
inter-residue contact information is present only for residues 42-60, the sequence is
designed based on inter-residue contact information as well as for amino acid secondary
structure propensities for this part only, while the rest of the sequence is designed for
amino acid secondary structure propensities. As Zhang and Skolnick’s potential contain
both inter-residue contact matrix and amino acid secondary structure propensity Tables,
we consider it suitable to design sequences in this stage. Hence, the energy as given by
Eq. 4.10 for sequence numbers 42-60 is,
( )60 60 60
42 42 42
1( , ) , ( )
2i j i
Total i j ij i j i
x x x
E C x x E x x E xβ= = =
= +∑ ∑ ∑ (6.1)
while for the rest of the sequence, the energy is given by,
{ }129
1
( ) 42 60i
Total i
x
E E x iα=
= ∉ −∑ (6.2)
Since, the conformation between positions 42-60 is specified, we consider the secondary
structure in this part also known, and hence in energy evaluation (Eq. 6.1) we consider
amino acid secondary structure propensities for β -strands for this part, while for the rest
of the sequence we use amino acid secondary structure propensities for α -helix only.
We generate 200 energy optimized sequences using the double sigmoid
formulation with Zhang and Skolnick’s potentials. The optimization is done using IPOPT
solver on a Xeon 3.0 GHz processor desktop computer which takes approximately an
hour to design a sequence. Since the wild-type sequence is assumed unknown, we prefer
not to use it to score the designed sequences. In such a situation, we adopt the following
approach. Since our structure prediction program cannot predict secondary structures, we
use two web-based servers, the GOR4 online server (http:// npsa-pbil.ibcp.fr / cgi-bin /
npsa_automat.pl ? page=npsa_gor4.html, Garnier et al., 1996), and the hierarchic neural
networks (HNN) online server (Guermeur, http:// npsa-pbil.ibcp.fr / cgi-bin /

119
npsa_automat.pl ? page=npsa_nn.html), to predict secondary structures from the designed
sequences (for a short discussion on these secondary structure prediction methods refer to
Appendix B4). We then match the secondary structures of the designed sequences
predicted by the two abovementioned secondary structure prediction servers with the
wild-type secondary structures (Fig. 6.2 b) and rank the designed sequences according to
maximum number of secondary structure matches. In view of lack of access to
experimental facilities to test our designed sequences, we consider the results from the
secondary structure prediction servers as a substitute for experimental results, which
would have decided the selection of best designed sequences. The secondary structure
prediction results using the GOR4 and HNN servers for the wild-type sequence of 1LZE
is shown in Fig. 6.3. A few of the highest ranking designed sequences are presented in
Fig. 6.4.
We select a few of the best-ranking sequences from the predictions of both GOR4
and HNN servers as candidates for ab initio structure prediction. We use Tozzini’s
parameters (Tozzini and Rocchia, 2006) to construct OB-CG models of α -helices
predicted by the GOR4 and HNN servers. The geometry of the backbone ( Cα atoms
only) in the range of residue numbers 42-60 is taken from the PDB file. The α -helices
and the fixed part of the conformation are then connected in order by coarse-grained
Fig. 6.3. The GOR4 and HNN servers’ secondary structure prediction results for the
wild-type sequence of 1LZE.

120

121
beads to construct an unfolded structure, which serves as initial input to the tertiary
structure prediction program. We consider first five to ten of the highest-ranking
sequences predicted by each server and generate an ensemble of unfolded conformations
in a manner similar to that described in the chapter 5. For each sequence, approximately
50-100 unfolded conformations are generated. Thus, we generate approximately 1000
conformations to search the conformation space. Next, we optimize these structures to
form an ensemble of energy optimized structures. Since, the number of conformations to
be optimized is quite large (approximately 1000 in number), we use the nonlinear
conjugate gradient method which uses the one-bead coarse-grained (OB-CG) model with
rigid secondary structures. We use both the coarse-grained energy potentials, i.e., our
continuous function formulation incorporating the MJ matrix (Eq. 5.1) and Levitt’s
potentials (Eqs. 5.7 and 5.8) to predict tertiary structures from the abovementioned
unfolded states. We show two such predicted structures in Fig. 6.5 (b and d). The
unfolded states from which these structures were achieved are also shown (Fig. 6.5 a and
c respectively). A few of the best optimized structures are presented in Table 6.1. The
average time taken for optimization using nonlinear conjugate gradient on a Xeon 3.0
GHz processor desktop computer with Levitt’s model is approximately one hour, whereas
for our continuous function formulation incorporating the MJ matrix it is approximately
6-10 minutes.
In Table 6.1, we show the results of both the energy models. Under each energy
model, the first column indicates the DRMSD of the unfolded structures from the native
conformation in the PDB file 1LZE. The second column indicates the DRMSD of the
conformation after optimization using the corresponding potentials. In Table 6.1, we use
Fig. 6.4. Few of the designed sequences having high secondary structure prediction
similarity with wild-type secondary structure of 1LZE. The corresponding server
name, i.e., either GOR or HNN is indicated at the top before the sequences and
corresponding secondary structure prediction results are shown. The number of
secondary structure matches are also indicated. The secondary structures are
indicated below the sequences. As before, h – alpha helix and e – beta strand.
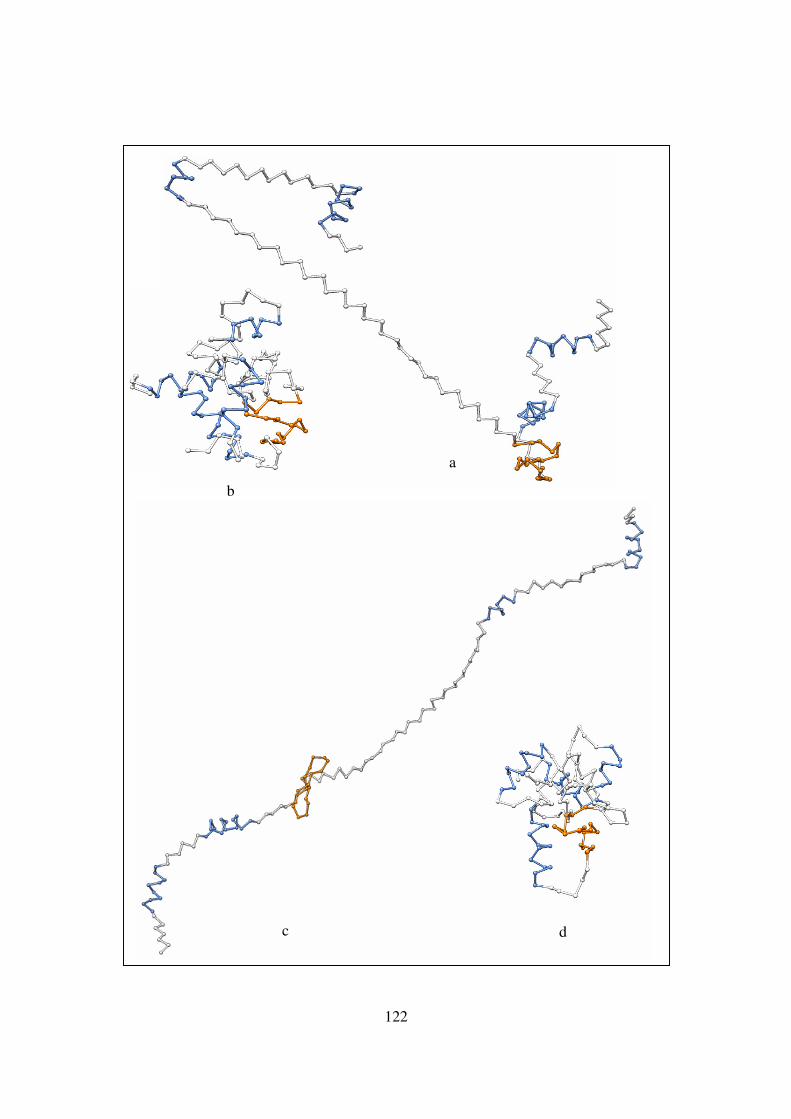
122
a
b
c d

123
a second metric apart from DRMSD, namely, the Template Modeling (TM) score (Zhang
and Skolnick, 2004, 2005). The TM-score is independent of the size of the protein and
can identify protein substructures (Zhang and Skolnick, 2004). Furthermore, recently it
has been claimed that the TM-score can be used for protein topology classification, i.e.,
“protein pairs with a TM-score >0.5 are mostly in the same fold while those with a TM-
score <0.5 are mainly not in the same fold” (Xu and Zhang, 2010). Thus, in the absence
of experimental verifications, the TM-score, apart from the DRMSD, can be considered
to be a suitable metric to rank our predicted tertiary structures. As shown in Table 6.1, we
achieved the highest TM-scores of 0.456 using Levitt’s potentials and 0.36 using our OB-
CG function from MJ matrix. The corresponding conformations are shown in figs. 6.5 b
and 6.5 d respectively. We consider the conformation shown in Fig. 6.4b as the best
conformation among all predicted conformations. This completes Step (iv) in our
simultaneous sequence and conformation search flow-diagram (Fig. 1.7).
In the next step, we design energy minimized sequences and further test them for
stability. Since, the tertiary structure is now available, we use the quadratic programming
method with atomistic potentials and amino acid composition constraints. Thus, in this
step we use the optimization function given by Eqs. 4.14 and 4.15. We generate Table
Fig.6.5. Tertiary structure prediction results using OB-CG model and rigid
secondary structures. The alpha-helices are colored blue; the part of the
conformation which we consider fixed, i.,e., backbone for residues 42-60 is colored
orange. The initial inputs to optimization are the conformations (a) and (c). The
final conformations after optimization are (b) and (c) respectively.
a) Initial conformation to optimization. The DRMSD from native state of 1LZE is
41.5 Å.
b) Final conformation after optimization from the structure shown in (a). The
DRMSD from native state of 1LZE is 7.52 Å.
c) Initial conformation to optimization. The DRMSD from native state of 1LZE is
46.2 Å.
d) Final conformation after optimization from the structure shown in (c). The
DRMSD from native state of 1LZE is 8.32 Å.

124
6.1. Few selected examples of tertiary structure prediction results using both energy
models. Under each energy model, the first column indicates the DRMSD of the unfolded
conformation from 1LZE ( C α− coordinates only) which serves as input to optimization
program. The second column is the DRMSD of the energy optimized structure from
1LZE ( C α− coordinates only). The third column shows the TM-score of the same
energy optimized structure. The conformation of the two highest TM-scoring structures
(0.456 and 0.36) are shown in figs. 6.4.b and 6.4.d respectively. The unfolded structures
from which these structures were achieved are shown in figs. 6.4.a and 6.4.c respectively.
OB-CG function from MJ Levitt
DRMSD
(Ǻ) form
native state
before
optimization
DRMSD
(Ǻ) form
native state
after
optimization
TM-score
DRMSD
(Ǻ) form
native state
before
optimization
DRMSD
(Ǻ) form
native state
after
optimization
TM-score
46.2 8.32 0.36 41.5 7.52 0.456
36.83 8.38 0.31 23.6 7.9 0.34
65.4 8.56 0.32 33.67 7.83 0.32
16.05 7.34 0.34 29.35 7.66 0.4
51.6 8.2 0.31 24.9 8.04 0.32
44.2 8.34 0.304 23.7 8.68 0.35
approximately 200 energy minimized sequences using the quadratic programming
method. The average time taken for designing each sequence by the IPOPT optimization
solver on a Xeon 3.0 GHz computer was approximately 10-12 hours. As mentioned
before, (step (vi) in Fig. 1.7), our aim now is to select the best sequence among these
designed sequences based on specificity requirements, i.e., the sequence which shows
maximum energy gap and minimum energy dispersion with respect to the selected

125
tertiary structure will be the best sequence. The tertiary structures which were not
selected as best designable structure can be used as decoy sets for checking specificity of
the designed sequences. As before (Chapter 4, the results section of the quadratic
programming method), we use the SCWRL software to determine rotamer conformations
and calculate energy using atomistic potentials. However, in calculating energies we
encounter a problem which we had not foreseen. Many of the coarse-grained tertiary
structures that we planned to use as decoy sets encounter some steric hindrance when the
designed sequences are threaded on them and energies are calculated. This happens as we
design the sequences using atomistic potentials, whereas the tertiary structures are
derived using coarse-grained energy models. If there are no steric hindrances, the energy
of the sequence is always a negative number. However, when there is a steric hindrance,
the energy of the sequence becomes a high positive number. Thus, when there are no
steric overlaps, the energy gap between two energy minimized conformations is less in
magnitude than the calculated energies. If there is steric hindrance in one of the
conformations, the calculated energies between the two conformations are opposite in
sign, and consequently the energy gaps are higher in magnitude than the corresponding
energies. If there is steric hindrance in both conformations, both conformations are
unsuitable for the sequence; however, one conformation is still selected as the suitable
one and competes with other low energy structures. We observed that such high energy
gaps are spurious in nature in the sense that they cause non-specificity of the designed
sequences and we had to abandon specificity check for the designed sequences using
atomistic potentials. We return to this issue in the discussion section.
Next, we use the coarse-grained energy model of Levitt (Levitt, 1976) to check
for specificity of the designed sequences. The energy gap versus dispersion of energies of
the designed sequences are presented in Fig. 6.6 a. The maximum ratio of the energy gap
between the selected structure and average on energies of all structures (decoy sets) to the
dispersion of energies is 0.121. The sequence with this ratio is shown in Fig. 6.6 b (the
bottom sequence). According to the specificity test (step (vi) in Fig. 1.7), this sequence is
the most specific and hence most suitable for the conformation shown in Fig. 6.4 b with
respect to the other conformations. We also align this sequence with the best sequence in
the previous iteration, i.e., the one based on which the most suitable conformation was

126
Fig. 6.6. a) Plots of energy gap (E target structure avg decoys
E E− −∆ = − ) versus Eσ (the
standard deviation of energy of the decoy set structures) for the designed sequences.
The target structure is the conformation shown in Fig. 6.4 b. The sequence that has
maximum /E Eσ∆ ratio is shown on the plot with the red dot.
b) The sequence with maximum /E Eσ∆ (bottom one) aligned with the best
designed sequence in the previous iteration (top one), i.e., the sequence for the
conformation in Fig. 6.4 b, using CLUSTAL (version 1.83) software.
(a)
(b)

127
achieved (Fig. 6.5 b). The determination of this sequence completes one iteration of our
simultaneous sequence and conformation search method (flowchart presented in Fig.
1.7).
6.4 Discussion
In the last section we presented one iteration in the loop of the flowchart of simultaneous
sequence and conformation search presented in Fig. 1.7. Before proceeding further, i.e.,
going through the second loop in the iteration which may take more than one month even
after our stress on computationally efficient techniques, one needs to re-evaluate the
computational techniques in the light of the results presented in the previous section. In
this section we evaluate the computational techniques and models in the order in which
the results were presented in the previous section. However, we do not question the
specified conditions, for example, how does one know beforehand the number of residues
or chains or the amino acid composition of the protein which is to be designed, which our
simultaneous sequence and conformation search technique requires as minimum inputs.
In the first stage we designed sequences using the double sigmoid method and
Zhang and Skolnick’s potentials. Although, the use of statistical potentials has been
questioned in designing sequences (Thomas and Dill, 1996), and methods have been
suggested for deriving statistical potentials based on the target backbone and decoy sets
(Mirny and Shakhnovich, 1996), we consider the use of Zhang and Skolnick’s potentials
in the first step justified, as minimal information is available at this stage in our problem,
viz. the length of the sequence and the backbone conformation for a limited part of the
chain. We had to consider a potential which does not depend on the tertiary structure
information, and a potential based on secondary structure propensities best suits this
purpose. However, once the sequences are designed, we do not rank them according to
energy, but with respect to some other criteria (like energy gap and energy dispersion).
Again, since no information on conformation for the whole sequence or decoy structures
is available, we could not apply the method of ranking the sequences as in Chapter 4.
Instead, we decided to rank the sequences in the order of predicted secondary structure
matches with the wild-type sequence. To this end, we used two online secondary
structure prediction servers, and considered the results from them as substitute to

128
experimental data. However, as Fig. 6.3 shows, the secondary structure prediction results
from these two servers differs from actual secondary structures of 1LZE even for the
wild-type sequence. Thus the deviations incurred in misprediction of secondary structures
will affect tertiary structure prediction, which in turn will affect sequence prediction in
the next round, and this effect will go on increasing. To the best of our knowledge we do
not know any secondary structure prediction technique that is 100% accurate, and at this
point, we are not certain how this will affect the convergence of results in the sequence
and conformation spaces.
In the second stage, we predicted tertiary structures from unfolded conformations
with rigid secondary structures. The underlying assumption of this prediction is that
secondary structures are formed before tertiary structures. However, as modern theories
suggest (Sinha and Udgaonkar, 2009), the secondary and tertiary structures evolve
simultaneously and the formation of one effects the other and vice-versa. Thus, in the
ideal case we should do structure prediction in one step, rather than in two steps.
However, even though we tried to develop models to predict secondary structures based
on continuous optimization techniques (which is presented as a short section in Chapter
5), a continuous function that can simultaneously develop secondary and tertiary
structures when minimized is yet to be developed. Furthermore, the coarse-grained
energy potentials based on which we do tertiary structure prediction, namely the MJ
matrix and Levitt’s potentials have their own limitations in reflecting the actual energy
governing protein folding. The limitations of theses coarse-grained energy models
became apparent when we threaded designed sequences on the energy minimized
structures and tried to calculate energy based on atomistic potentials. Many of the
conformations, which occupy a minimum energy position in the energy-conformation
space encountered steric hindrances and consequently high energy when different
sequences were threaded onto them. The reason for this may be that, coarse-grained
potentials, being lower degree polynomials than their atomistic counterparts, allow the
residues to be packed together closer than is actually possible, and the extent of packing
grows with the size of the protein. In such a situation, we think the best remedy is to run
some fine-grained atomistic potential-based simulation such as molecular dynamics on

129
each of the tertiary structures derived using coarse-grained potentials. But that would
involve substantial computation power and time.
As an alternative, we tried the other option; we threaded the sequences and calculated
energy using coarse-grained potentials. The reader may question the test for specificity
using coarse-grained energy where the sequences were designed using atomistic
potentials. We agree that such a question is justified. However, as indicated in the
motivation section of Chapter 1, there are few examples of protein design using search in
both sequence and conformation spaces. We were motivated to find out whether the
question of convergence in sequence and conformation spaces can be asked at all with the
methods that we have developed. Thus, although we have used different potentials to
design sequence and check for specificity, all the blue colored dots in Fig. 6.6(a) indicate
that our simultaneous sequence and conformation search strategy indeed yield sequences
that show specificity of the designed target tertiary structure over other competitive
structures, even though marginally. Thus, the aim that we set to achieve is a feasible one
and methods that we developed show some promise.
6.5 Closure
In this chapter, we considered the re-design of an existing enzyme, the hen egg-white
lysozyme (PDB ID 1LZE) using the sequence design and conformation search tools that
we develop in the previous chapters. We briefly described the target protein and how we
model the re-design of the same protein. We next applied the simultaneous sequence and
conformation search strategy that we proposed in Chapter 1 and presented the results in a
stepwise manner as described in our method. We closed this chapter with a discussion on
the efficacy of the computational tools in the light of the results.

130
7. Towards parallelization of tertiary structure
prediction using Graphics Processor Unit (GPU)
based parallel computation
• We give a brief introduction and our motivation for GPU-based code for tertiary
structure prediction.
• We describe the logic of conversion of the CPU-based code to the GPU-based
code.
• We present a case study with CPU and GPU based codes and present the results.
• We close this chapter with a summary.
7.1 Introduction and motivation
In this chapter we describe our attempt to parallelize the tertiary structure prediction
computer program using Graphics Processor Unit (GPU) based parallel programming
computation model, the Compute Unified Device Architecture (CUDA) introduced by
NVIDIA in 2007 (Kirk and Hwu, 2010). We select the tertiary structure prediction
program over the sequence design program as the optimization solver (nonlinear
conjugate gradient) for tertiary structure prediction is coded in-house. The nature of the
tertiary structure prediction program further makes it suitable for parallel computation in
the following way.
The tertiary structure prediction program optimizes the free energy of the protein
molecule calculated as a function of the coordinates of the residues in the OB-CG model
(Eq. 5.4). Thus if there are N residues in a protein, there are ( )N○ variables. Here we
assume that after reducing the secondary structures as rigid bodies, there is no reduction
in the order of the number of optimization variables, which is the case for the proteins
considered in this work. While calculating the non-bonded interaction energies (Eq. 5.1
or 5.7), for every residue, its interaction energy with all other residues are calculated.
Thus, for the non-bonded interaction energy there are ( )2N○ calculations (one may
consider a reduced number of calculations based on a cut-off list of interacting residues,
but since there is large change in conformation during optimization, such a cut-off list

131
will have to be updated from time to time during optimization). Gradient-based
optimization requires the calculation of the gradient of the optimizing function with
respect to the design variables (Eq. 5.5) during every step in optimization. Thus, if the are
N variables in an optimization program, the gradient will contain N components that will
involve ( )N○ calculations. Hence, the gradient of the non-bonded interaction energy
will involve ( )3N○ calculations. Apart from the non-bonded energy, the bond length
and bond-angle constraints each individually involve ( )N○ calculations. Thus, their
gradients will individually involve ( )2N○ calculations. A typical example of tertiary
structure optimization presented in the last chapter involved 210 design variables and
8,128 non-bonded interactions. Thus, the gradient of the non-bonded energy will involve
8,128 210 1,706,880× = order of numerical calculations. Even though we coded our
tertiary structure prediction programs in C++ which is compiled using the Intel C++
compiler with O3 optimization options and employed on a 64 bit Xeon 3.0 GHz
computer with static memory allocation to minimize time for allocation and de-allocation
of memory, it still takes more than an hour on the average to run each simulation. The
main reason for this is that in the CPU based code, the large number of calculations
involved in calculating energies and their gradients are done sequentially through loops
(for or while), although each of these calculation is independent of one another (Fig.
7.1). Hence, much time can be saved if each of the energy calculations and the
corresponding gradients can be done in parallel. The ability of CUDA to handle large
number of numerical computations in parallel was the motive behind our effort to convert
the CPU based codes into GPU based codes.
Our GPU programs were run on a C1060 Tesla Cluster with CUDA driver
version 3.20. Our CPU programs were run on an Intel i7 2.67 GHz processor.
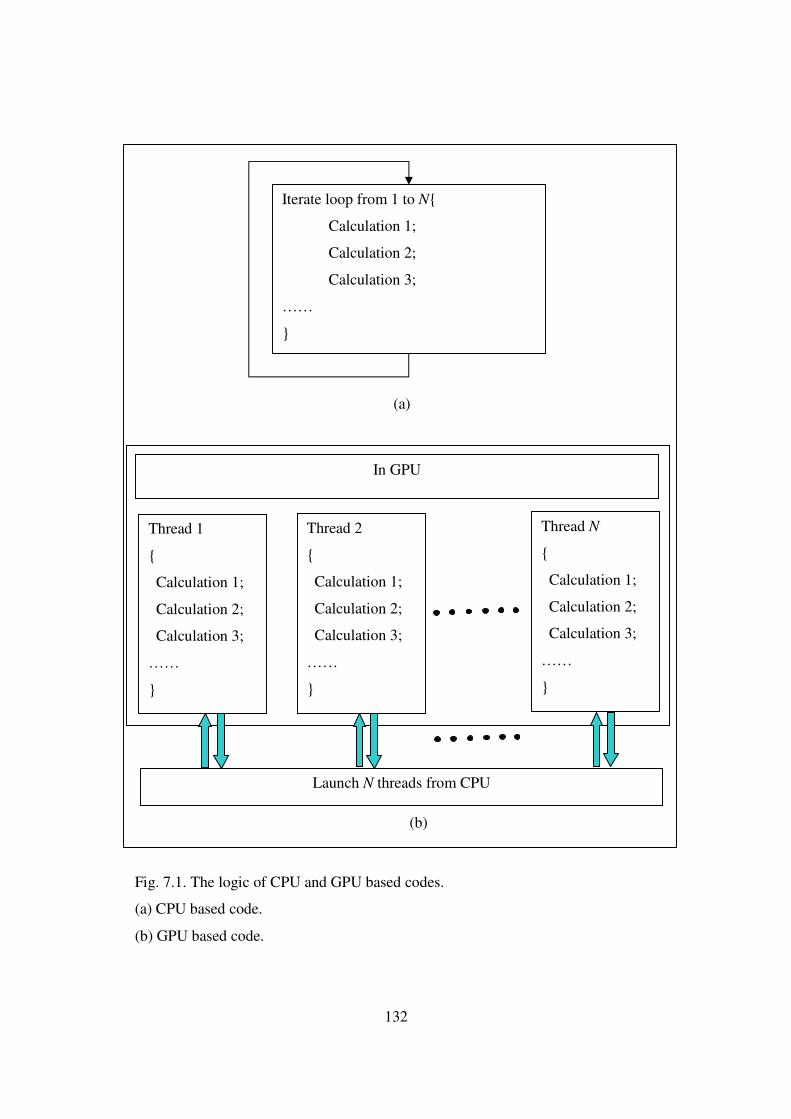
132
Iterate loop from 1 to N{
Calculation 1;
Calculation 2;
Calculation 3;
……
}
(a)
Fig. 7.1. The logic of CPU and GPU based codes.
(a) CPU based code.
(b) GPU based code.
Thread 1
{
Calculation 1;
Calculation 2;
Calculation 3;
……
}
Thread 2
{
Calculation 1;
Calculation 2;
Calculation 3;
……
}
Thread N
{
Calculation 1;
Calculation 2;
Calculation 3;
……
}
Launch N threads from CPU
In GPU
(b)

133
7.2 From CPU-based code to GPU-based code
To understand the GPU-based computer code one has to understand the CPU-based
computer code. The flow diagram of our tertiary structure prediction code is presented in
Fig. 7.2. First, the input data for calculations are read; the input data consist of initial co-
ordinates of the protein molecule and its amino acid sequence, the secondary and fixed
structures information which are treated as rigid bodies during optimization, the reference
coordinates with respect to which the rotation of rigid bodies are determined (generally
these are the co-ordinates of the secondary and fixed structure from the PDB file of the
protein), and energy parameters (MJ matrix or Levitt’s van der Waals’ and solvation
energy parameters). Next, the residues are classified into rigid variables and free
variables. The residues which belong to secondary or fixed structure are classified as
rigid residues. The other residues are classified as free residues. The coordinates of the
free residues form the optimization variables X . Also within X are the coordinates of
Fig. 7.2. Flow diagram of tertiary structure prediction code.
Read data: initial co-ordinates, amino acid sequence, reference co-ordinates, energy
parameters, secondary structure information
Write data: final energy, final co-ordinates
Nonlinear Conjugate Gradient performs optimization using
,X
E E ∇ = evalE_dEdx ( ),miscellaneous dataX
Classify the residues as part of rigid bodies and free; the co-ordinates of the free residues form the optimization
variables X

134
the first and last residues in a secondary structure which determines its position during
optimization and with respect to which the co-ordinates of the other residues inside that
secondary structure are calculated (Fig. 5.16). The optimization is done using the
nonlinear conjugate gradient algorithm which is presented in Appendix #. The nonlinear
conjugate gradient algorithm calls the function evalE_dEdx which calculates the bond
length (Eq. 5.3), the bond angle (Eq. 5.2) and the non-bonded (Eq. 5.1 or Eqs. 5.7 and
5.8) energies as functions of X and also the gradient of the same energies with respect to
X . As mentioned before, it is the calculation of the energies and their derivatives which
require maximum computational effort and is the target of parallelization; the other
functions, namely, reading data, classifying residues into free and rigid ones and
determining X , the nonlinear conjugate gradient, and writing functions are same for the
CPU and GPU-based codes, and is done on the CPU.
Let us now describe the function evalE_dEdx which is central to our
computation. The algorithm of evalE_dEdx is presented in Fig. 7.3. Each step in
evalE_dEdx can be done on the GPU. However, whenever a GPU based function is
called, some latency time is involved in transfer of data from CPU to GPU and back (ref.
CUDA manuals). If the number of calculations are small, then it is more efficient to do
the calculations on the CPU. Hence, a limit on the number of iterations for a specific
calculation which determines whether it will be done on the GPU or the CPU is
necessary; in our case, we choose this limit as 28 = 256. Thus, if the number of iterations
is less than 256, we do it on CPU; if greater, we do it on GPU. For the proteins on which
we have worked in this thesis, all the derivative calculations and non bondedE − fall in the
range of GPU, the rest are done on CPU. We next present the results for the GPU and
CPU based codes for a test case.

135
[ ,dE
Ed X
] = evalE_dEdx ( X , residueID, N , refX , RBdata, sequence, parametersE ){
1. Determine residue specific coordinates { }, ,x y z from design variable X
and derivatives of the same with respect to design variables.
( )1 if ( ) coordinate and ( ) variable represent the same coordinate
( )
in the same residue
0 otherwise
dx ix i X j
dX j=
=
2. Determine position of rigid bodies ( RBV ) by calculating the translation vector
transV and the rotation angle θ of RBV with respect to reference coordinates refX .
Calculate the rotation matrix ℜ for each rigid body. Calculate
, and transdV d d
d X d X d X
θ ℜ as follows,
{ } { }{ }
( )
( )
( )
11 2
2 11 2
2
1 2
( ) , , , ,
, ,( )
cos see Eq. 5.2
1
1
trans i i i ref ref ref
trans i i i
i
i i
RB
V i x y z x y z
d x y zdV i
d X d X
u u
dxd du duu u
dx dxd X d Xu u
d d d
dd X d X
V i V
θ
θ
θ
θ
−
= −
=
= •
−= • + •
− •
ℜ ℜ=
= ( )
( ) ( )
trans ref
RB trans
i V
dV i dV i d
d X d X d X
+ ℜ
ℜ= +
3. Calculate the position of the residues inside the rigid bodies using transV and ℜ .
Also, calculate their derivatives with respect to X .
4. Calculate bond length energy, bondedE (Eq. 5.3) and its derivatives with respect to
X , bondeddE
d X.

136
7.3 A case study with CPU and GPU based codes
We now consider an example for which we run both our CPU and GPU based codes and
see their performances. This example is an unfolded structure of hen egg-white lysozyme
(PDB ID 1LZE) with 129 residues in which the residue positions 42-60 are kept fixed
throughout optimization, i.e., they act like boundary conditions or constraints. There are
six secondary structures which are considered as rigid bodies during optimization. After
omitting the fixed part and considering the secondary structures as rigid bodies this
problem consist of 219 design variables. In Table 7.1 we show the time required by CPU
and GPU for different calculations in the algorithm of evalE_dEdx which is shown in
Fig. 7.3. In this table, we show results for those calculations for which the number of
iterations per function call of evalE_dEdx were greater than 256, for that is the limit
above which we do the calculation in the GPU as mentioned before.
(continued from previous page)
5. Calculate bond angle energy, Eθ (Eq. 5.2) and its derivatives with respect to X ,
dE
d X
θ .
6. Calculate nonbonded energy, (Eq. 5.1 or Eqs. 5.7 and 5.8) non bondedE − and its
derivatives with respect to X , non bondeddE
d X
− .
7. Calculate the total energy totalE (Eq. 5.4) and its derivatives totaldE
d X.
}
Fig. 7.3. Flowchart of the algorithm evalE_dEdx.
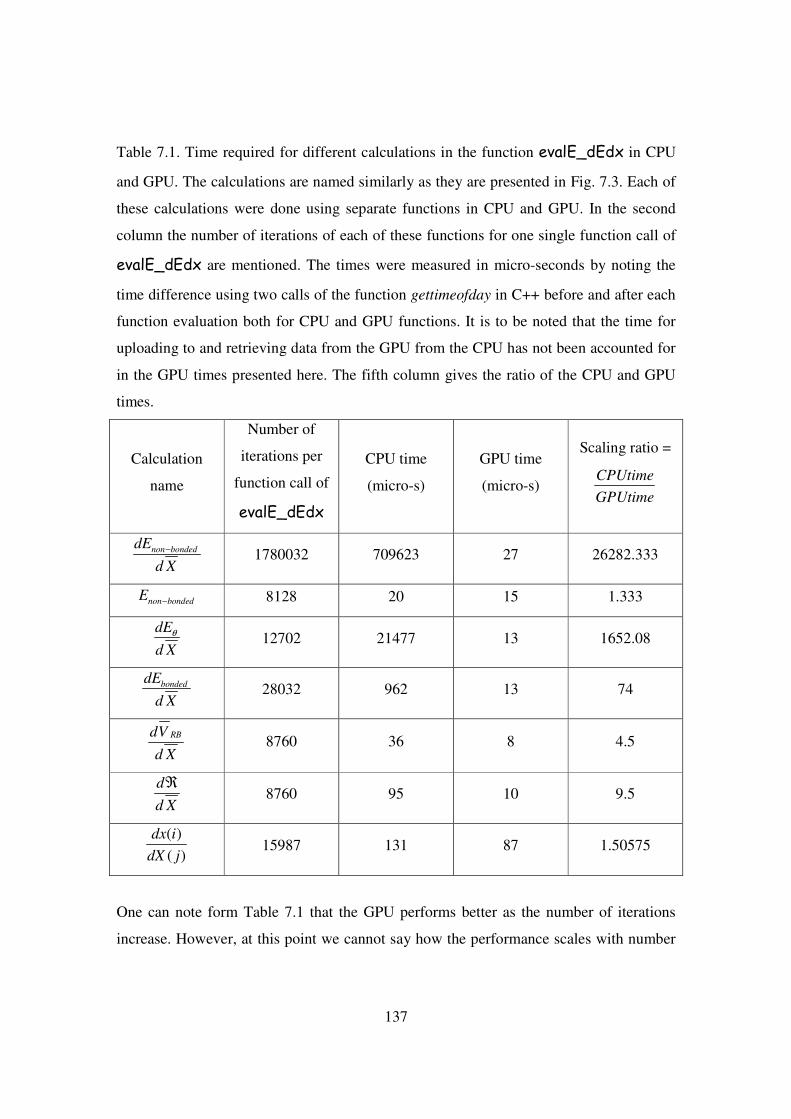
137
Table 7.1. Time required for different calculations in the function evalE_dEdx in CPU
and GPU. The calculations are named similarly as they are presented in Fig. 7.3. Each of
these calculations were done using separate functions in CPU and GPU. In the second
column the number of iterations of each of these functions for one single function call of
evalE_dEdx are mentioned. The times were measured in micro-seconds by noting the
time difference using two calls of the function gettimeofday in C++ before and after each
function evaluation both for CPU and GPU functions. It is to be noted that the time for
uploading to and retrieving data from the GPU from the CPU has not been accounted for
in the GPU times presented here. The fifth column gives the ratio of the CPU and GPU
times.
Calculation
name
Number of
iterations per
function call of
evalE_dEdx
CPU time
(micro-s)
GPU time
(micro-s)
Scaling ratio =
CPUtime
GPUtime
non bondeddE
d X
− 1780032 709623 27 26282.333
non bondedE − 8128 20 15 1.333
dE
d X
θ 12702 21477 13 1652.08
bondeddE
d X 28032 962 13 74
RBdV
d X 8760 36 8 4.5
d
d X
ℜ 8760 95 10 9.5
( )( )
dx i
dX j 15987 131 87 1.50575
One can note form Table 7.1 that the GPU performs better as the number of iterations
increase. However, at this point we cannot say how the performance scales with number

138
of iterations. It appears to depend on the nature of calculations. For example, both RBdV
d X
and d
d X
ℜ are iterated 8760 times; however, in the evaluation of
d
d X
ℜ the GPU is much
faster than the CPU than in the evaluation of RBdV
d X. There is another interesting point
which we would like to mention. It appears that the formula for all energy evaluation and
their derivatives pose complex tasks for the GPU. When we compiled with –arch=sm_13
option (required for double precision calculations) we got a failure message for all energy
and corresponding derivative function evaluations stating that memory and register
requirements were too high for all these functions. When we compiled with the
debugging option –g –G along with –arch=sm_13 option, the memory and register
requirements of these functions came down and we were able to run our program.
However, we are not sure as to how the –g –G option affects the performance of GPU2..
However, even with the –g –G option (about which we are not sure how it affects
the performance of the GPU) it leaves no doubt from the performance of the GPU as
presented in Table 7.1 that if we use the GPU based functions, the overall performance of
the optimization program (Fig. 7.2) will be much better. However, when we ran all our
inputs in the GPU based code, which amounts to a total of approximately 1000 initial
conformations, only in two cases the optimization code based on GPU actually converged
whereas, the optimization code based on CPU converges for greater than 99% cases. To
give the reader a feeling of how the CPU and GPU based codes converge to the final
result we present the results of iterations in Fig. 7.4. In this figure, we print the function
value and the norm of the gradient of the same for the function evalE_dEdx after every
219 (the number of variables in the chosen optimization problem) steps in optimization
using the nonlinear conjugate optimization program. Since the nonlinear conjugate
gradient algorithm consists of the linear conjugate gradient algorithm which is iterated
unless the function value or the gradient of the same converges, we chose the number of
2 Upon posting a message in the NVIDIA website as to why such a thing happens, the only reply received stated that our energy evaluation and derivative formulas were too complex to be handled in the GPU.

139
GPU code CPU code
continued on the next page…

140
GPU code CPU code
Fig. 7.4. The function value and norm of the gradient of the same for evalE_dEdx
for the CPU and GPU codes. The function value and the norm are printed after
every 219 (the number of variables in the chosen optimization problem) steps in
optimization (indicate by “iter” in the figure) using the nonlinear conjugate
optimization program. This also corresponds to one complete iteration of the linear
conjugate gradient part in the nonlinear conjugate optimization program (see
Appendix B3). Due to lack of space we have not presented the result every 219
iterations in this figure, but with arbitrary gaps indicated by dashed lines. For the
full figure enlisting the results for both the GPU and CPU codes the reader is
requested to refer to Appendix C.

141
variables in optimization as a suitable step for printing values as it corresponds to the end
of every linear conjugate gradient solution to the nonlinear optimization problem. For a
comprehensive overview of the results, we do not present the full run in Fig. 7.4, but
insert gaps which are indicated with dashed lines in the same figure. The interested reader
is requested to refer to Appendix C for a step by step comparison of the performance of
the GPU and CPU codes.
We gave the same inputs and same set of energy calculation parameters to both
the CPU and GPU codes. At the start (iteration number 0), the results diverge by
55.41 10 %−× in function value and 54.4 10 %−× in the norm of the gradient, which we
ascribe due to the difference of the compilers, namely nvcc for the GPU code, and g++
for the CPU code. However, by 219 iterations we notice that the results for GPU and
CPU codes diverge by 2.41% in function evaluation and 10.75% in the norm of the
gradient. Towards the end, i.e., after 19272 iterations it appears that the GPU and CPU
codes have settled to their own, but slightly different converged values. However, we
notice that for a large number of iterations, i.e., from 19272 to 27375, i.e., for 8103 steps
the GPU code has run with small changes in the function value and oscillatory changes in
the gradient of the function which is indicated by its norm. This has lead to lower
performance of the GPU code which converged in 8058 s compared to the CPU code
which converged in 7560 s. We would also like to mention here that in the majority of
cases in which the GPU code failed (998 out of 100) as compared to the CPU code which
succeeded in 99% cases, the optimization in GPU showed such oscillatory nature of
gradient evaluations before failure in optimization. We did not investigate this matter any
further.
7.4 Closure
In this chapter we presented our efforts of parallelizing the tertiary structure prediction
program using GPU based CUDA programming model. We described our motivation for
going from CPU to GPU for tertiary structure prediction and present the logic of
converting the CPU-based code to the GPU-based code. We took up a test case and
present the times taken by various calculations in the CPU and the GPU. We also
presented the result of optimization for the GPU and CPU codes in steps so that their
performances can be compared.

142
8. Closure and Future Work
• The work presented in the thesis is summarized and its conclusions are noted.
• Contributions of this thesis are presented.
• Based on the conclusions, a few directions for future work are proposed.
8.1 Summary and Conclusions
The preceding chapters presented the computational methods that we developed and the
results that we achieved in our goal of functionalistic protein design using continuous
optimization methods. From our approach, it should be clear to the reader that wherever
possible, we have tried to formulate problems as continuous functions so that gradient-
based optimization techniques can be used. However, in a few instances (e.g., secondary
structure prediction) we have not been successful so far, and these problems can be taken
up as part of future work.
Protein design is an unsolved problem. Although questions have been raised in
the past on the feasibility of protein design, ingenious experiments have proved it to be
possible. However, theoretical models that can provide a sound basis for protein design
are yet to be developed. Even the atomistic potentials are questionable after a certain
limit of approximation, as they are not derived by solving Schrödinger’s wave equation,
as should be done in case of true atomistic potentials. So, the computational techniques
that are employed in protein design employ some form of knowledge-based parameters to
some extent or other, for example, from more or less generic atomistic potentials derived
from experimental results of small molecules to fully knowledge based statistical
potentials designed for specific proteins. Because of so much dependence on knowledge
derived from experiments over time, the computational techniques which are used for
protein design are almost always heuristic. However, we have tried to the best of our
efforts to adhere to computational methods that have a sound mathematical framework,
namely, gradient-based continuous optimization.
Computational protein design has sometimes been labeled as an N-P hard
problem, implying that it is intractable from theoretical and computational point of view.
Hence, we took a cautious approach when we set the goal of protein design and devised a
method of reducing the amino acid set from 20 to a much lower number for sequence

143
design. However, ultimately we did not use reduced amino acid sets in sequence design
as we came to learn and use IPOPT, a gradient-based optimization solver which can
handle nonlinear optimization problems with a large number of optimization variables
and nonlinear set of constraints. The sequences designed using IPOPT based on free
energy minimization were tested on two different uncorrelated design criteria different
from ours, namely, match with wild-type sequences and check for stability by calculating
the Z-score based on decoy sets. In the other domain of functionalistic protein design,
namely, structure prediction, we concentrated on coarse-grained models as fine grained
models can make the folding funnel of the designed protein rugged. With this view, we
developed our own continuous coarse-grained function from the Miyazawa-Jernigan inter
residue contact energy matrix. However, our efforts towards ab initio structure prediction
faced problems as the coarse-grained potentials developed were not conducive to
secondary structure formation. Keeping the larger goal of tertiary structure prediction
from sequence in view, we used web based servers for secondary structure prediction as
an intermediate step and went ahead with our own model of tertiary structure prediction
using continuous optimization. The tertiary structure prediction program was used with
two different coarse-grained energy models and validated on several proteins. We would
like to mention here that we have not been successful to formulate an optimization
function which can successfully pair up beta strands to form beta sheets which determine
the tertiary structure of proteins where beta sheets are present. The validations we
performed were only on alpha-helical proteins.
Once the tools were developed, we combined them to form a computational
strategy of simultaneous search in sequence and conformation spaces for functionalistic
protein design. The selection of a target protein was difficult as we have no resource of
experimental techniques or expertise. We chose the re-design of a well studied protein,
the hen egg-white lysozyme, which has a functionally active site and two key residues
that form the enzyme-substrate complex which can serve as the structural and functional
constraints in our optimization model. Although the tertiary structure of the hen egg-
white lysozyme is stabilized by four disulphide bonds and we had not taken into account
the formation of disulphide bonds in our optimization function formulation, we decided
to go ahead and see the structure prediction results with the tertiary structure prediction

144
program that we have developed. The structural constraint which we imposed was a part
of that part of the lysozyme structure dominated by beta sheets, and hence our selection
of that part was more guided by computational constraints than by actual functional
constraints of the same protein.
Following the simultaneous sequence and conformation search strategy proposed,
we first design sequences based on statistical potentials using only that part of the
structure specified as a functional constraint. At this point, a difficult situation arose
because of lack of access to experimental verification. The experimental results are much
needed feedback for rectifying computational models as the knowledgeable reader will
know. We could not also use the wild-type sequence as that would fail our purpose of re-
design of the hen egg-white lysozyme. One can argue that we could rank the designed
sequences in terms of their energy and select a small set possessing the lowest energies.
However, as our previous results on sequence design demonstrated a few limitations of
statistical potentials, we sought to devise a different criterion for selecting the designed
sequences based on maximum predicted secondary structure matches with the secondary
structures of lysozyme using web-based secondary structure prediction servers. With the
best ranked sequences selected as per the new design criterion, we did tertiary structure
prediction from approximately 1000 unfolded structures using two different coarse-
grained energy models and selected the structure with best TM-score. Following the
simultaneous sequence and conformation search strategy, we again designed sequences
for the selected tertiary structure, but now we used atomistic potentials and imposed
amino acid composition constraints to ensure stability of the designed sequences. This
time we test the designed sequences using the Z-score criterion with the unqualified set of
predicted teriary structures in the previous round serving as decoy sets for the designed
sequences. In this thesis we demonstrated two rounds of sequence prediction and one
round of structure prediction in the simultaneous sequence and conformation search
procedure with satisfactory results despite our lack in experimental feedback.
The thesis ends with an effort to parallelize the tertiary structure prediction code
using GPU based CUDA programming. The next section shows a bulleted list of
contributions of the work presented in this thesis.

145
8.2 Contributions of the Thesis
• Conception, formulation, and application of a simultaneous sequence and
conformation search method aimed at computational design of de novo functional
proteins.
• Development of a novel method of amino acid grouping using MMDS on the MJ
matrix and determination of a best sets of reduced amino acid alphabets.
• Formulation of protein sequence design for a fixed backbone as a free energy
minimization problem using a novel double sigmoid function, its application
using different potentials, and its verification on four different proteins.
• Formulation of protein sequence design for fixed backbone as a free energy
minimization problem using atomistic potentials and its verification on four
different proteins.
• Formulation of a novel continuous function one-bead coarse-grained model for
protein structure prediction from the MJ matrix.
• Formulation of a coarse-grained energy function for the formation of helices
using continuous optimization.
• Development of a novel coarse-grained elastic network model for ab initio protein
structure prediction and its validation using proteins of different sizes.
• Development of coarse-grained protein tertiary structure prediction model using
rigid secondary structures and its validation using proteins of different sizes.
• Coupling the above tertiary structure prediction program with on-line secondary
structure prediction servers for ab initio protein structure prediction.
• Formulation of a novel algorithm for simultaneous sequence and conformation
space searches using some of the aforementioned sequence and conformation
search tools.
• Exploring the parallelizing protein tertiary structure prediction program using the
GPU-based CUDA programming technique.

146
8.2 Future Work
As stated earlier, the work done in this thesis is the beginning of a long-term goal that
needs to be pursued over time. The scope of functionalistic protein design is enormous
from user-specific drug design to re-engineering microorganisms to produce biofuels and
myriad chemicals in environment friendly manner; in short, it can usher a new revolution
with the hope of a greener world. The aim of computational techniques is to aid
researchers in designing the right sort of experiments from the ad infinitum complexity
inherent in biological phenomena. Furthermore, as stressed previously, we sought to take
a separate path than conventional computational approaches by adopting computationally
efficient techniques.
A good way to identify future tasks is to critically review our own computational
tools. Let us ask the question that might have already come to the reader’s mind: when it
is already known that both the sequence and conformation space search that this thesis
proposes are non-convex problems, why are we sticking to continuous optimization that
only gives a local minimum? To this our answer has always been and still is that we stand
by continuous optimization because of its computational efficiency. However, if a
function has multiple minima, then the results of continuous optimization will definitely
depend on the starting points. To this end, we have always used random initial starting
points. Given the nonlinear nature of optimization function and the associated constraints
for sequence and conformation search, and admitting that we do not know beforehand the
intervals in which the minima lie, sampling techniques that can efficiently search the
design spaces and provide suitable initial inputs to continuous optimization are necessary.
Coming to sequence design, the pair-wise potential technique certainly has
drawbacks, which was perhaps reflected when we used the Z-score based design criterion
to test the designed sequences. Any formulation that eliminates the pair-wise potential
approach for calculating the implicit solvation energy will not only aid our attempt using
gradient based optimization formulations for sequence design, but also to the
computational de novo protein design research community in general. In the present work
we incorporated matching of predicted secondary structures using web-based secondary
structure servers as a criterion for selecting designed sequences as we were not confident
on the statistical potentials we used. However, if we had designed statistical potentials

147
based on the type of biomolecule that we were about to design, then we could have
trusted the designed sequence ranking on the basis of energy alone. Thus, the design of
statistical potentials using gradient based optimization methods and formulations can be
another development in our model.
In the structure prediction area, a lot of improvements can be made. First,
consider the prediction of secondary structures. We tried to develop a continuous
function formulation for prediction of helices and strands, but it failed. This can be a
good work in future. We also faced the problem of beta strand pairing to form beta
sheets. This is an open problem, and to the best of our knowledge there are only a few
works in this direction, that too, using other methods such as global optimization and
machine learning techniques. In tertiary structure prediction, we used the Cartesian
coordinates of the residues as variables in optimization. However, internal coordinates
such as dihedral angles may be used in tertiary structure prediction. The use of dihedral
angles will simplify the incorporation of fine-grained potentials in energy formulation.
However, structure prediction using dihedral angles has been done using stochastic
methods such as Monte-Carlo, and we found that formulating energy in terms of dihedral
angles makes the optimization function more nonsmooth which in turn affects the
performance of continuous optimization. Thus, the formulation of energy in terms of
dihedral angles and atomistic potentials which can be optimized using the nonlinear
conjugate gradient program that we have used can be another welcome development in
our method.
After designing sequences in the second round of simultaneous sequence and
conformation search we had to use coarse-grained potentials for testing the designed
sequences for stability even though they were designed using atomistic potentials because
of lack of some fine-tuned simulation, i.e., equilibrating the coarse-grained energy
minimized structures using MD simulations. We have recently come to know that CUDA
enabled MD simulations, for example, GROMACS, have become available, which are
much faster than CPU-based MD simulations. Such MD simulations can be used to
equilibrate the large number (we had approximately 1000) of coarse-grained energy
minimized structures before we thread the designed sequences on them and calculate the
Z-score.

148
Insisting on using continuous optimization methods, incidentally, motivates the
development of rigorous principles and models in protein design. This, we believe,
should be pursued with equal vigor in conjunction with heuristic and stochastic methods
largely followed in the current literature in the field. Generality of the methods–another
aspect emphasized in this work–rather than specific methods for specific proteins, helps
bring protein design into the ambit of rigorous methods that the engineering fields enjoy
today.

149
Appendix A
Alanine Ala A
Methionine Met M
Glycine Gly G
Valine Val V
Leucine Leu L
Isoleucine Ile I
Cystine Cys C
Lysine Lys L
Arginine Arg R
Asparagine Asn N
Histidine His H
Phenylalanine Phe F
Proline Pro P
Serine Ser S
Threonine Thr T
Tyrosine Tyr Y
Tryptophan Trp W
Glutamine Gln Q
Aspartic acid Asp D
Glutamic acid Glu E

150
Fig. A1. All twenty amino acids with their full name, the structure, followed by three-letter abbreviated name and single letter code. Black dots represent Carbon, red Oxygen, blue Nitrogen, yellow Sulphur and white Hydrogen.

151
Appendix B
B.1 Interior Point Optimization (IPOPT)
Interior Point Optimization (IPOPT) is based on the barrier function methods, a class of
gradient based continuous optimization techniques, which bypass the problem of
identifying the active set of constraints that occurs during the solution of constrained
quadratic programming problems by introducing a barrier function in the objective
function for optimization (Wächter, 2002). The barrier function method used in IPOPT
optimizes the following nonlinear optimization function defined by
( )
. . ( ) 0
0
nx
Min f x
s t c x
x
∈
=
≥
�
(B.1)
as a series of approximate solutions for a sequence of barrier problems defined as
( )( )
1
( ) : ( ) ln
. . ( ) 0
n
ni
xi
Min x f x x
s t c x
µϕ µ∈
=
= −
=
∑� (B.2)
for a decreasing sequence of barrier parameters µ converging to zero (Wächter and
Biegler, 2006). IPOPT takes the primal-dual approach in which the dual variables
defined by
( )( )
:i
iv
x
µ= (B.3)
( { }1,2,....,i I n∈ ⊆ is the set of indices for the bounded variables) are incorporated into
the KKT equations as
( ) ( )
( ) ( ) 0
( ) 0
0 for i i
f x c x v
c x
x v i I
λ
µ
∇ + ∇ − =
=
− = ∈
(B.4)
For 0µ = Eq. (B.4) along with the inequalities ( ) 0ix ≥ and ( ) 0iv ≥ are equivalent to the
KKT conditions for Eq. B.1 (Wächter, 2002). IPOPT computes an approximate solution
to the barrier problem (Eq. B.2) for a fixed value of the barrier parameter µ , then
decreases µ and continues the solution of the next barrier problem from the approximate
solution of the previous one (Wächter and Biegler, 2006). The algorithm consists of two

152
loops, an “outer loop” in which the approximate solution of the barrier problem (Eq. B.2)
satisfies a given tolerance, and an “inner loop” in which Eq. B.4 is solved. IPOPT
converges superlinearly under standard second order sufficiency conditions for the
problem defined in Eq. B.1. For more details on the algorithm and the package, the reader
is requested to consult the IPOPT homepage (www.coin-or.org/Ipopt/) and the following
references (Wächter, 2002, Wächter and Biegler, 2006).
B.2 SCWRL
SCWRL is a side-chain prediction program based on a graph theoretic algorithm. The
graph-theory algorithm is based on representing side chains as vertices in an undirected
graph. Residues having non-zero rotamer interactions energies are considered to have an
edge between the vertices in the graph. Thus, the protein with its rotamers having
interactions with one another can be considered as a big undirected graph. SCWRL uses a
backbone-dependant rotamer library (Dunbrack and Karplus, 1994, Dunbrack and Cohen,
1997, Dunbrack, 2002), an energy function based on the log probabilities of the rotamers
in the library and a repulsive steric energy term. The newer versions of SCWRL (versions
3.0 and above) uses a biconnected graph partitioning algorithm which breaks the large
graph representing the protein into smaller components and then using DEE to solve
(Goldstein, 1994) the best rotamer conformations for each of the smaller components.
The interested reader is requested to refer to the details of the algorithm in the paper by
Dunbrack’s group (Canutescu et al., 2008).
B.3 Nonlinear conjugate gradient method
The conjugate gradient method was first proposed by Hestenes and Stiefel (Hestenes and
Stiefel, 1952) for solving a system of linear equations. Later Fletcher and Reeves
(Fletcher and Reeves, 1964) extended the conjugate gradient method for optimizing
general nonlinear functions. Conjugate gradient methods have low memory requirements
compared to other unconstrained convex optimization solvers like the quasi-Newton
methods (Bazara et al., 1993), and takes at most n iterations to solve an unconstrained
convex problem of n variables.
Let ( )f x be the function to be optimized. We used the nonlinear conjugate
gradient algorithm from the freely available text by Jonathan Richard Shewchuk

153
(www.cs.cmu.edu/~quake-papers/painless-conjugate-gradient.pdf) with minor
modifications, for example, using the Polak-Rebierre formula for β calculation. The
algorithm is presented below.
( ) ( )
( ) ( )
{ }
0;
, _ ;
_ ;
;
( & & & & )
{
, _ ;
;
;
( 0; ; )
{
( 0)
new
old old
new
old
new new old
old old
old
old
iter
f f evalE dEdx x
f Big number
r f
While iter Maxiter r f f
f f evalE dEdx x
d f
r d
for i i N i
if iter
else
δ ε
σ δ
=
∇ =
=
= −∇
< < − <
∇ =
= −∇
=
= < + +
== =
{ }
( ) ( )
_ ( , );
;
, _ ;
;
;
;
;
;
}
1;
}
new new
new new
new old
new
old old
new
old new
line search x d
x x d
f f evalE dEdx x
r f
r r r
r r
r r
d r d
r r
iter iter
σ α
α
α
β
β
=
=
= +
∇ =
= −∇
∆ = −
∆=
= +
=
= +
i
i
In the above algorithm, evalE_dEdx is the function that takes in the vector X (which we
denote by x for simplicity) and returns the function value f and the gradient f∇ evaluated
at X , and N is the size of X . The line_search function calculates α using the following
formula,

154
( )( ) ( )
f x d
f x d d f x dα σ
σ
∇= −
∇ + − ∇
i
i i (B.5)
The parameters , and Maxiter δ ε have values 8 6 31.0 10 , 1.0 10 and 1.0 10− −× × ×
respectively.
B.4 Online Secondary Structure prediction servers
A comprehensive list of online secondary structure prediction servers is given at
http://www.expasy.ch/tools/#secondary. From this website we selected two online
secondary structure prediction servers, namely, the GOR4 secondary structure prediction
server (http://npsa-pbil.ibcp.fr/cgi-bin /npsa_automat.pl?page=npsa_gor4.html, Garnier et
al., 1996), and the HNN server (Guermeur, http://npsa-pbil.ibcp.fr/cgi-bin/
npsa_automat.pl?page=npsa_nn.html) for predicting secondary structures from
sequences.
The GOR method uses the information function defined by,
( | )
( ; ) log( )
P S RI S R
P S
=
(B.6)
where, S is one of the three conformations, i.e. alpha helix (H), beta strand (E) or coil (C),
and R is one of the 20 amino acids. Thus, ( | )P S R is the conditional probability for
observing a conformation S when a residue R is present given by
( | ) ( , ) / ( )P S R P S R P R= , and ( )P S is the probability of observing S. For a large
database with known sequences and secondary structures the above probabilities can be
calculated as ,( , ) /S RP S R f N= , ( ) /RP R f N= , and ( ) /SP R f N= , where N is the total
number of amino acids in the database, ,S Rf is the frequency of residues R observed in
the conformation S in the same database, Rf is the total number of residues R, and
Sf the
total number of residues observed in the conformation S in the same database. Thus,
( )( )
, /( ; ) log
/S R R
S
f fI S R
f N
=
(B.7)
The actual program incorporates corrections for levels of data. For details of the program,
the interested reader is requested to refer to the paper by Robson and co-workers Garnier
et al., 1996).

155
The HNN method (Guermeur, PhD thesis) is based on an ensemble method based on a
multivariate linear regression algorithm which finds estimates of the class posterior
probabilities using optimization and generalized Vapnik-Chernonekis dimensions.

156
Appendix C
Simulation results for the GPU (left) and CPU (right) based tertiary structure prediction
codes. The time is measured in seconds. The function value and the norm are printed
after every 219 (the number of variables in the chosen optimization problem) steps in
optimization (indicate by “iter”) using the nonlinear conjugate optimization program.
This also corresponds to one complete iteration of the linear conjugate gradient part in the
nonlinear conjugate optimization program (see Appendix B3).

157

158

159

160

161

162

163
References
1. Abe, H., and Gō, N., Noninteracting local-structure model of folding and
unfolding transition in globular proteins. II. Application to two-dimensional
lattice proteins, Biopolym., 1981, vol. 20, p.p. 1013-1031.
2. Abkevich, V. I., Gutin, A. M., and Shakhnovich, E. I., Improved design of stable
and fast-folding model proteins. Fold. Desg., 1996, vol. 1, p.p. 221-230.
3. Alvizo, O., and Mayo, S. L., Evaluating and optimizing computational protein
design force fields using fixed composition-based negative design. PNAS, 2008,
vol. 105(34), p.p. 12242-12247.
4. Ananthasuresh, G. K., Protein sequence design on the basis of topology
optimization techniques. IUTAM symposium on topology design optimization of
structures, machines and materials, Ed. Bendsøe, Olhoff N. and Sigmund O.,
Springer, 2006, p.p. 237-248.
5. Anfinsen, C. B., et al., The kinetics of formation of native ribonuclease during
oxidation of the reduced polypeptide chain. PNAS, 1961, vol. 47(9), p.p. 1309-
1314.
6. Anfinsen, C. B., Principles that Govern the Folding of Protein Chains. Science,
1973, vol.181 (4096), p.p. 223-230.
7. Ashby, M. F., and Johnson, K., Materials and Design: The Art and Science of
Material Selection in Product Design, 2002, © Butterworth-Heinemann.
8. Arora, N., and Jayaram, B., Strength of hydrogen bonds in alpha helices. J.
Comput. Chem., vol. 18, no. 9, pp. 1245-1252, 1997.
9. Atilgan, A. R., et al., Anisotropy of fluctuation dynamics of proteins with an
elastic network model. Biophy. J., 2001, vol. 80, p.p. 505-515.
10. Aydin, Z., Altunbasak, Y., and Erdogan, H., Bayesian models and algorithms for
protein beta-sheet prediction. IEEE Trans. Comput. Biol. Bioinfo., 2011, vol. 8,
p.p. 395-409.
11. Ball, K. D., et al., The elastic net algorithm and protein structure prediction. J.
Comput. Chem., 2001, vol. 23, p.p. 77-83.

164
12. Bahar, I., Atilgan, A. R., and Erman, B., Direct Evaluation of thermal fluctuations
in proteins using a single-parameter harmonic potential. Fold. Des., 1997, vol. 2,
p.p. 173-181.
13. Bahar, I., and Jernigan, R. L., Vibrational dynamics of transfer RNAs:
Comparison of the free and synthetase-bound forms. JMB, 1998, vol. 281, p.p.
871-884.
14. Bahar, I., and Rader, A. J., Coarse-grained normal mode analysis in structural
biology. Curr. Op. Struct. Biol., 2005, vol. 15, p.p. 586-592.
15. Baker, D., An exciting but challenging road ahead for computational enzyme
design. Prot. Sci., 2010, vol. 19, p.p. 1817-1819.
16. Bazara, M. S., Sherali, H. D., and Shetty, C. M., Nonlinear Programming Theory
and Algorithms, 2nd Ed., © John Wiley & Sons, Inc.
17. Bernstein, F. C., Koetzle, T.F., Williams, G. J. B., Meyer, Jr. E. F., Brice, M. D.,
Rodgers, J. R., Kennard, O., Shimanouchi, T., and Tasumi, M., The protein data
bank: a computer based archival file for macromolecular structures. JMB, 1977,
vol. 112, p.p. 535-542.
18. Bolon, D. N., and Mayo, S. L., Enzyme-like proteins by computational design.
PNAS, 2001, vol. 98, p.p. 14274-14279.
19. Bowie, J. U., Lüthy, R., and Eisenberg, D., A method to identify protein
sequences that fold into known three-dimensional structure. Science, 1991, vol.
253, p.p. 164-170.
20. Bradley, P. et al, TRILOGS: discovery of sequence-structure patterns across
diverse proteins. PNAS, 2002, vol. 99, p.p. 8500-8505.
21. Brooks, B., and Karplus, M., Harmonic dynamics of proteins: Normal modes and
fluctuations in bovine pancreatic trypsin inhibitor. PNAS, 1983, vol. 80, p.p.
6571-6575.
22. Brown, S., Fawzi, N. J., and Head-Gordon, T., Coarse-grained sequences for
protein folding and design. PNAS, 2003, vol. 100(19), p.p. 10712-17.
23. Bryngelson, J. D., et al., Funnels, pathways, and the energy landscape of protein
folding: a synthesis. Proteins, 1995, vol. 21, p.p. 167-195.

165
24. Bryson, J. W. et al, From Coiled Coils to Small Globular Proteins: Design of a
Native-Like Three-Helix Bundle. Prot. Sci., 1998, vol. 7, p.p. 1404-1414.
25. Butterfoss, G. L., and Kuhlman, B., Computer-based design of novel protein
structures. Ann. Rev. Bioph. Biomol. Strs., 2006, vol. 35, p.p. 49-65.
26. Cannata, N., et al., Simplifying amino acid alphabets by means of a branch and
bound algorithm and substitution matrices. Bioinformatics, 2002, vol. 18, p.p.
1102-1108.
27. Canutescu, A. A., Shelenkov, A. A., and Dunbrack Jr., R. L., A graph-theory
algorithm for rapid protein side-chain prediction. Prot. Sci., 2003, vol. 12, p.p.
2001-2014.
28. Case, D. A., et al., The Amber biomolecular simulation programs. J. Comput.
Chem., 2005, vol. 26, p.p. 1668-1688.
29. Chan, H. S., Folding alphabets. Nature Struct. Biol., 1999, vol. 6 (11), p.p. 994-
996.
30. Chen, J., et al., Increase in the thermostability of the staphylococcal nuclease:
implications for the origin of protein thermostability. JMB, 2000, vol. 303, p.p.
125-130.
31. Chin, T. M., Berndt, K. M., and Yang, N. C., Self-Assembling Hexameric Helical
Bundle Forming peptides. JACS, vol. 114, p.p. 2279-2280.
32. Chiu, T-L., and Goldstein, R. A., Optimizing potentials for inverse protein folding
problem. Prot. Engg., 1998, vol. 11(9), p.p. 749-752.
33. Chotia, C., Principles that Determine the Structure of Proteins. Ann. Rev.
Biochem., 1984, vol53, p.p. 537-572.
34. Cieplak, M., et al, Amino acid classes and protein folding problem. J. Chem.
Phys., 2001, vol. 114, p.p. 1420-1423.
35. Cohen, V. and Parry, D., Alpha-Helical Coiled Coils and Bundles: How to Design
Alpha-Helical protein. Proteins, 1990, vol. 7, p.p. 1-15.
36. Cook, R. D., Malakus, D. S., Plesha, M. E., and Witt, R. J., Concepts and
applications of finite element analysis; Wiley, 2002 edition.

166
37. Cornell, W. D., et al., A second generation force field for the simulation of
proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc., 1995, vol. 117,
p.p. 5179-5197.
38. Cornette, J. L., et al., Hydrophobicity scales and computational techniques for
detecting amphipatic structures in proteins. J. Mol. Biol., 1987, vol. 195, p.p. 659-
685.
39. Dahiyat, B. I., and Mayo, S. L., Protein design automation. Prot. Sci., 1996, vol.
5, p.p. 895-903.
40. Dahiyat, B. I., and Mayo, S. L., De novo protein design: fully automated sequence
selection. Science, 1997a, vol. 278, p.p. 82-87.
41. Dahiyat, B. I., and Mayo, S. L., Probing the role of packing specificity in protein
design. PNAS, 1997b, vol. 94, p.p. 10172-10177.
42. Dahiyat, B. I., Sarisky, C. A., and Mayo, S. L., De novo protein design: towards
fully automated sequence selection. JMB, 1997, vol. 273, p.p. 789-796.
43. Dantas, G., et al., A large scale test of computational protein design: Folding and
stability of nine completely redesigned globular proteins. JMB, 2003, vol. 332,
p.p. 449-460.
44. Das, R., and Baker, D., Macromolecular modeling with Rosetta. Ann. Rev.
Biochem., 2008, vol. 77, pp. 363-382.
45. David, S. R., et al, Functional Rapidly Folding Proteins from Simplified Amino
Acid Sequences. Nat. Struct. Biol., 1997, vol. 4(10), p.p. 805-809.
46. Dayhoff, M. O., Eck, R. V., Park, C. M., A model of evolutionary change in
proteins. Atlas of protein sequence and structure (Ed. M. O. Dayhoff , Maryland:
National Biomedical Research Foundation), 1972, p.p. 89-100.
47. DeGrado, Design of peptides and proteins. Adv. Prot. Chem., 1988, vol. 39, p.p.
51-124.
48. De Grado, W. F., and Lear, J. D., Conformationally constrained alpha-helical
peptide models for protein ion channels, Biopoly., 1990, vol. 29, p.p. 205-213.
49. DeGrado, W. F., Raleigh, D. P., and Handel, T., De novo protein design: what are
we learning ?. Curr. Op. Struct. Biol., 1991, vol. 1, p.p. 984-993.

167
50. DeGrado, W. F., Summa, C. F., Pavone, V., Nastri, F., and Lombardi, A., De
Novo design and Structural Characterization of Proteins and Metalloproteins.
Ann. Rev. Biochem., 1999, vol. 68, p.p. 779-819.
51. Deisenhofer, J., Crystallographic refinement and atomic models of a human FC
fragment and its complex with fragment B of protein A from staphylococcus
areus at 2.9 and 2.8 angstroms resolution. Biochem., 1981, vol. 20, p.p. 2361-
2370.
52. Delarue, M., and Sanejouand, Y.-H., Simplified normal mode analysis of
conformational transitions in DNA-dependant polymerases: the elastic network
model. JMB, 2002, vol. 320, p.p. 1011-1024.
53. Delarue, M., and Dumas, P., On the use of low-frequency normal modes to
enforce collective movements in refining macromolecular structural models.
PNAS, 2004, vol. 101, p.p. 6957-6962.
54. DeMaeyer, M., et al., All in one: a highly detailed rotamer library improves both
accuracy and speed in the modeling of side chains by dead-end elimination. Fold.
Des., 1997, vol. 2, p.p. 53-66.
55. Derrida, B., Random energy model: limit of a family of disordered models. PRL,
1980, vol. 45(2), p.p. 79-82.
56. Desjarlais, J. R., and Handel, T. M., De novo design of the hydrophobic cores of
proteins. Prot. Sci., 1995, vol 4, p.p. 5803-5807.
57. Desjarlais, J. R., and Handel, T. M., Side-chain and backbone flexibility in protein
core design. JMB, 1999, vol. 289, p.p. 305-318.
58. Desjarlais, J. R., and Clarke, N. D., Computer Search Algorithms in Protein
Modofication and Design. Curr. Op. Struct. Biol., 1998, vol. 8, p.p. 471-475.
59. Desmet, J., De Maeyer, M., Hazes, B. and Lasters, I., The dead-end elimination
theorem and its use in protein side-chain positioning. Nature, 1992, vol. 356, p.p.
539-542.
60. Deutsch, J. M., and Kurosky, T., New algorithm for protein design. PRL, 1996,
vol. 76 (2), p.p. 323-326.
61. Dill, K. A., Theory for the folding and stability of globular proteins, . Biochem.,
1985, vol. 24, p.p. 1501-1509.

168
62. Dill, K. A., Dominant Forces in protein Folding. Biochem., 1990, vol. 29 (31),
p.p. 7133-7155.
63. Dill, K. A., and Chan, H. S., From Levinthal to pathways to funnels. Nat. Struct.
Biol., 1997, vol. 4 (1), p.p. 10-19.
64. Dokholyan, N. V., What is protein design alphabet ?. Prot. Struct. Fun. Bioinfo.,
2004, vol. 54, p.p. 622-628.
65. Doruker, P., Jernigan, R. L., and Bahar, I., Dynamics of large proteins through
hierarchical levels of coarse-grained structures. J. Comput. Chem., 2002, vol. 23,
p.p. 119-127.
66. Drexler, K. E., Molecular engineering: An approach to the development of
general capabilities for molecular manipulation. PNAS, 1981, vol. 78(9), p.p.
5275-5278.
67. Dunbrack Jr., R. L., and Karplus, M., Conformational analysis of the backbone-
dependant rotamer preferences of protein sidechains. Nat. Struct. Biol., 1994, vol.
1, p.p. 334-340.
68. Dunbrack Jr., R. L., and Cohen, F. E., Bayesian statistical analysis of protein side-
chain rotamer preferences. Prot. Sci., 1997, vol. 6, p.p. 1661-1681.
69. Dunbrack Jr., R. L., Rotamer libraries in 21st century. Curr. Op. Struct. Biol.,
2002, vol. 12, p.p. 431-440.
70. Dwyer, M. A., Looger, L. L., and Hellinga, H. W., Computational Design of a
Biologically Active Enzyme. Science, 2004, vol. 304, p.p. 1967-1971.
71. Eichinger, B. E., Elasticity theory I. Distribution function for perfect phantom
networks. Macromol., 1972, vol. 5, p.p. 496-505.
72. Erman, B. , Bahar, I., and Jernigan, R. L., Equilibrium states of rigid bodies with
multiple interaction sites: Application to protein helices. J. Chem. Phys., 1997,
vol. 107, no. 6, pp. 2046-2058.
73. Erman, B., and Dill, K., Gaussian model of protein folding. J. Chem. Phys., 2000,
vol. 112(2), p.p. 1050-1056.
74. Farinas, E., and Regan, L., The de novo design of a rubredoxin-like Fe site. Prot.
Sci., 1998, vol. 7, p.p. 1939-1946.

169
75. Fletcher, R., and Reeves, C., Function minimization by conjugate gradients,
Computer J., 1964, vol. 7, p.p. 149-154.
76. Flory, P. J., Statistical thermodynamics of random networks. Proc. Roy. Soc. A.,
1976, vol. 351, p.p. 351-380.
77. Floudas, C. A., Fung, H. K., McAllister, H. R., Mönnigmann, M., and Rajgaria,
R., Advances in protein structure prediction and de novo protein design: a review.
Chem. Engg. Sci., 2006, vol. 61, p.p. 966-988.
78. Fraternali, F., and van Gunsteren, W. F., An efficient mean solvation force model
for use in molecular dynamics simulations of proteins in aqueous solution. JMB,
1996, vol. 256, p.p. 939-948.
79. Fung, H. K., Welsh, W. J., and Floudas, C. A., Computational de novo peptide
and protein design: Rigid templates versus flexible templates. Ind. Eng. Chem.
Res., 2008, vol. 47, p.p. 993-1001.
80. Garnier, J., et al., GOR secondary structure prediction method version IV.
Methods in Enzymology, 1996, Ed. Doolittle, R. F., vol. 266, p.p. 540-553.
81. Ghadiri, M. R., et al., Self-assembling organic nanotubes based on a cyclic
peptide architecture. Nature, 1993, vol., 366, p.p. 324-327.
82. Gillespie, B., et al., NMR and temperature junp measurements of de novo
designed proteins demonstrate rapid folding in the absence of expilict selection
for kinetics. JMB, 2003, vol. 330, p.p. 813-819.
83. Gibney, B. R. et al, Synthesis of novel proteins. Curr. Op. Chem. Biol., 1997, vol.
1, p.p. 537-542.
84. Gō, N., Noguti, T., and Nishikawa, T., Dynamics of small globular proteins in
terms of low-frequency vibrational modes. PNAS, 1983, vol. 80, p.p. 3690-3700.
85. Goffe, W. L., Ferrier, G. D., and Rogers, J., Global optimization of statistical
functions with simulated annealing. J. Econometrics, 1994, vol. 6, p.p. 65-99.
86. Goldstein, R. F., Efficient rotamer elimination applied to protein side chains and
related spin glasses. Biophys. J., 1994, vol. 66, p.p. 1335-1340.
87. Gordon, D. B., and Mayo, S. L., Radical performance enhancements for
combinatorial optimization algorithms based on the dead-end elimination
theorem. J. Comput. Chem., 1998, vol. 19(13), p.p. 1505-1514.

170
88. Gordon, D. B., Marshall, S. A., and Mayo, S. L., Energy functions for protein
design. Curr. Op. Struct. Biol., 1999, vol. 9, p.p. 509-513.
89. Gordon, D. B., and Mayo, S. L., Branch-and-Terminate: a combinatorial
optimization algorithm for protein design. Structure, 1999, vol. 7, p.p. 1089-1098.
90. Grove, A., et al., A molecular blueprint for the pore-forming structure of voltage-
gated ion channels, PNAS, 1991, vol. 88, p.p. 6418-6422.
91. Guermeur, Y., Combinaison de classifieurs statistiques, Application a la
prediction de structure secondaire des proteins. PhD Thesis. (http://npsa-
pbil.ibcp.fr/NPSA/npsa_references.html#hnn)
92. Güner, U., Arkun, Y., and Erman, B., Optimum folding pathways of proteins:
their determination and properties. J. Chem. Phys., 2006, vol. 124, 139411(1-12).
93. Gutte, B., et al.,
94. Halilgolu, T., Bahar, I., and Erman, B., Gaussian dynamics of folded proteins.
PRL, 1997, vol. 79(16), p.p. 3090-3093.
95. Halilgolu, T., et al., How similar are protein folding and protein binding nuclei?
Examination of vibrational motions of energy hot spots and conserved residues.
Biophys. J., 2005, vol. 88, p.p. 1552-1559.
96. Hao, M-H, and Scheraga, H. A., Designing potential energy functions for protein
folding. Curr. Op. Struct. Biol., 1999, vol. 9, p.p. 184-188.
97. Harbury, P. B., et al, High-Resolution Protein Design with Backbone Freedom.
Science, 1998, vol. 282, p.p. 1462-1467.
98. He, J., et al., Efficiently explore the energy landscape of proteins in molecular
dynamics simulations by amplifying collective motions. Biophys. J., 2003, vol.
119, p.p. 4005-4017.
99. Hecht, M. H., et al, De Novo Design, Expression, and Characterization of Felix:
A Four-Helix Bundle Protein of Native-Like Sequence. Science, 1990, vol. 249,
p.p. 884-891.
100. Hecht, M. H., De novo design of beta-sheet proteins. PNAS, 1994, vol. 91, p.p.
8729-8730.
101. Hecht, M. H., Strategies for the Design of Novel Proteins. Protein Engineering
and Design, 1996, Academic Press Inc., Ed. Carey, P. R., p.p. 1-46.

171
102. Hedstrom, L., Converting trypsin to chymotrypsin: the role of surface loops,
Science, 1992, vol. 255, p.p. 1249-1253.
103. Heinkoff, S., and Heinkoff, J. G., Amino acid substitution matrices from protein
blocks. PNAS, 1992, vol. 89, p.p. 10915-10919.
104. Hellinga, H. W., et al., Construction of ligand binding sites in proteins of known
structure. II: Grafting of a buried transition metal binding site into E.Coli
thioredoxin. JMB, 1991, vol. 222, p.p. 787-803.
105. Hellinga, H. W., and Richards, F. M., Optimal sequence selection in proteins of
known structure by simulated evolution. PNAS, 1994, vol. 91, p.p. 5803-5807.
106. Hellinga, H. W., Rational protein design: Combining theory and experiment.
PNAS, 1997, vol. 94, p.p. 10015-10017.
107. Hess, B., et al, GROMACS 4: Algorithms for Highly Efficient, Load-Balanced,
and Scalable Molecular Simulation. J. Chem. Theory & Comput., 2008, vol.
4(2), pp. 435-447.
108. Hestenes, M. R., and Stiefel, E., Methods of conjugate gradients for solving
linear systems, J. Res. National Bereau of Standards, 1952, vol. 49, p.p. 409-
436.
109. Higgins, D. G., and Sharp, P. M., CLUSTAL: a package for performing multiple
sequence alignment on a microcomputer. Gene, 1988, vol. 73, p.p. 237-244.
110. Hill, T. L., An Introduction to Statistical Thermodynamics, © Dover, 1986.
111. Ho, S. P. , and DeGrado, W. F., Design of a 4-Helix Bundle Protein: Synthesis
of Peptides which Self-Associate into a Helical Protein. JACS, 1987, vol. 109,
p.p. 6751-6758.
112. Hodges, R. S. et al, Synthetic Model Proteins: Contribution of Hydrophobic
Residues and Disulphide Bonds to Protein Stability. Pep. Res., 1990, vol. 3, p.p.
123-137.
113. Holland, J. H., Adaptation in natural and artificial systems. 1992, © MIT press.
114. Horton et al, Principles of Biochemistry, 4th Ed. © Pearson Education Inc.
115. Irbäck, A., Peterson, C., Potthast, F., and Sandelin, E., Monte Carlo procedure
for protein design. PRL E, 1998, vol. 58 (5), p.p. 5249-52.

172
116. Inaka, K., et al., Crystal structures of the apo- and holomutant human lysozymes
with an introduced Ca2+ binding site. J. Biol. Chem., 1991, vol. 266, p.p.
20666-20671.
117. Jaenicke, R., et al., Conformation of a synthetic 34-residue polypeptide that
interacts with nucleic acids. FEBS Lett., 1980, vol. 114, p.p. 161-164.
118. Jernigan, R. L., and Bahar, I, Structure-derived potentials and protein
simulations. Curr. Op. Struct. Biol., 1996, vol. 6, p.p. 195-209.
119. Jha, A. N., Ananthasuresh, G. K., and Vishveshwara, S., Protein sequence
design based on the topology of the native state structure. J. Theo. Biol., 2006,
vol. 248, p.p. 81-90.
120. Jha, A. N., Ananthasuresh, G. K., and Vishveshwara, S., A search for energy
minimized sequences of proteins. PLOS 1, 2009, vol. 4(8), e6684.
121. Jiang, L., et al, De novo computational design of retro-aldol enzymes. Science,
2008, vol. 319, p.p. 1387-1391.
122. Johnson, R. A., and Wichern, Applied Multivariate Statistical Analysis, 2006, ©
Pearson Education Inc.
123. Keller, D. A., et al., Finding global minimum: a fuzzy end elimination
implementation. Prot. Engg., 1995, vol. 8, p.p. 893-904.
124. Kim., M. K., Chirikjian, G. S., and Jernigan, R. L., Elastic models of
conformational transitions in macromolecules. J. Mol. Graph. Model., 2002, vol.
21, p.p. 151-160.
125. Kim, M. K., Jernigan, R. L., and Chirikjian, G. S., An elastic network model of
HK97 capsid maturation, J. Struct. Biol., 2003, vol. 143, p.p.107-117.
126. Kirk, D. B., and Hwu, W., Programming Massively Parallel Processors A
Hands-on Approach, 2010, © Elsevier Inc.
127. Kissinger, C. R., et al., Crystal structure of an engrailed homeodomain/DNA
complex at 2.8 angstroms resolution: a framework for understanding
homeodomain/DNA interactions. Cell, 1990, vol. 63, p.p. 579-590.
128. Klauser, S., et al., Structure-function studies of designed DDT-binding
polypeptides. Biochem. Biophys. Res. Comm., 1991, vol. 179, p.p. 1212-1219.

173
129. Klepeis, J. L., and Floudas, C. A., ASTRO-FOLD: A combinatorial and global
optimization framework for ab initio prediction of three-dimensional structures
of proteins from amino acid sequences. Biophys. J., 2003, vol. 85, p.p. 2119-
2146.
130. Klepeis, J. L., and Floudas, C. A., Prediction of β -sheet topology and
disulphide bridges in polypeptides. J. Comput. Chem., vol. 24, pp. 191-208,
2003.
131. Koehl, P., and Delarue, M., Application of self-consistent mean-field theory to
predict proteins side-chains conformation and estimate their conformational
entropy. JMB, 1994, vol. 239, p.p. 249-275.
132. Koehl, P., and Levitt, M., De novo protein design. I. In search of stability and
specificity. JMB, 1999a, vol. 293, p.p. 1161-1181.
133. Koehl, P., and Levitt, M., De novo protein design. II. Plasticity in sequence
space. JMB, 1999a, vol. 293, p.p. 1183-1193.
134. Koehl, P., and, Levitt, M., Improved recognition of native-like protein structures
using a family of designed sequences. PNAS, 2002, vol. 99(2), p.p. 691-696.
135. Koh, S. K., Ananthasuresh, G. K. and Vishveshwara S., A deterministic
optimization approach to protein sequence design using continuous models. Int.
J. Rob. Res., 2005, vol. 24, p.p. 109-130.
136. Koh, S. K., Ananthasuresh, G. K., and Croke, C., A quadratic programming
formulation for the design of reduced protein models in continuous sequence
space. J. Mech. Des., 2005, vol. 127, p.p. 728-734.
137. Koh, S. K., Guangjun, L., and Zhu, W-H, A continuous protein design model
using artificial power law in topology optimization. J. Mech. Des., 2009, vol.
131, 041001.
138. Koisol, C., Goldman, N., and Buttimore, H. N., A new criteria and method for
amino acid classification. J. Theo. Biol., 2004, vol. 228, p.p. 97-106.
139. Kono, H., and Doi, J., Energy minimization method using automata network for
sequence and side-chain conformation prediction from given backbone
geometry. Proteins, 1994, vol. 19, p.p. 244-255.

174
140. Kono, H., and Saven, J. G., Statistical theory for protein combinatorial libraries.
Packing interactions, backbone flexibility and sequence variability of a main-
chain structure. JMB, 2001, vol. 306, p.p. 607-628.
141. Kraemer-Pecore C. M., et al, Computational Protein design, Curr. Op. Chem.
Biol., 2001, vol. 5, p.p. 690-695.
142. Krebs et al., Normal mode analysis of macromolecular motions in a database
framework: developing mode concentration as a useful classifying statistic.
Proteins, 2002, vol. 48, p.p. 682-695.
143. Kruskal J. B., and Wish M., Multidimensional Scaling,1978, Sage publications.
144. Kuhlman, B., O’Neill, J. W., Kim, D. E., Zhang, K. Y., and Baker, D., Accurate
computer-based design of new backbone conformation in the second turn of
protein L. JMB, 2002, vol. 315, p.p. 471-477.
145. Kuhlman, B., Dantas, G., Ireton, G. C., Varani, G., Stoddard ,B. L., and Baker,
D., Design of a novel globular protein fold with atomic-level accuracy. Science,
2003, vol. 302, p.p. 1364-1368.
146. Kundu, S., et al., Automatic domain decomposition of proteins by a Gaussian
network model. Proteins, 2004, vol. 57, p.p. 725-733.
147. Kundu, S., and Jernigan, R. L., Molecular mechanism of domain swapping in
proteins: an analysis of slower motions. Biophys. J., 2004, vol. 86, p.p. 3846-
3854.
148. Kurkcuoglu, O., et al., Mixed levels of coarse graining of large proteins using
elastic network model succeeds in extracting the slowest motions. Polym., 2004,
vol. 45, p.p. 649-657.
149. Kuroki, R., et al., Design and creation of a Ca2+ binding site in human
lysozyme to enhance structural stability. PNAS, 1989, vol. 86, p.p. 6903-6907.
150. Larson et al., Thoroughly sampling sequence space: Large-scale protein design
of structural ensembles. Prot. Sci., 2002, vol. 11, p.p. 2804-2813.
151. Lassila, J. K., Conformational diversity and computational enzyme design. Curr.
Op. Chem. Biol., 2010, vol. 14, p.p. 676-682.

175
152. Lasters, I., De Maeyer, M. and Desmet, J., Enhanced dead-end elimination in the
search for global minimum energy conformation of a collection of protein
sidechains. Prot. Engg., 1995, vol. 8, p.p. 815-822.
153. Lazar, et al., De novo design of the hydrophobic core of ubiquitin. Prot. Sci.,
vol. 6, p.p. 1167-1178.
154. Lee, C., and Levitt, M., Accurate prediction of the stability and activity effects
of site-directed mutagenesis on a protein core. Nature, 1991, vol. 352, p.p. 448-
451.
155. Lee C., Predicting protein mutant energetics by self-consistent ensemble
optimization. JMB, 1994, vol. 236, p.p. 918-939.
156. Leo-Macais et al., An analysis of core deformations in protein superfamilies.
Biophys. J., 2005, vol. 88, p.p. 1291-1299.
157. Lerner, R. A., et al., At the crossroads of chemistry and immunology: catalytic
antibodies. Science, 1991, vol. 252, p.p. 659-667.
158. Levitt, M., and Warshel, A., Computer simulation of protein folding. Nature,
1975, vol. 253, p.p. 694-698.
159. Levitt, M., A Simplified Representation of Protein Conformations for rapid
Simulation of Protein Folding. JMB, 1976, vol. 104, p.p. 59-107.
160. Levitt, M., Protein Folding by Restrained Energy Minimization and Molecular
Dynamics. JMB, 1983, vol. 170, p.p. 723-764.
161. Levitt, M., Sander, C., and Stren, P. S., Protein normal mode dynamics: Trypsin
inhibitor, Crambin, Ribonuclease and Lysozyme. JMB, 1885, vol. 181, p.p. 423-
447.
162. Li, H., Tang, C., and Wingreen, N. S., Nature of driving force for protein
folding: A result from analyzing statistical potential. Phy. Rev. Lett., 1997, vol.
79(4), p.p. 765-768.
163. Li, G., and Cui, Q., A coarse-grained normal mode approach for
macromolecules: an efficient implementation and application to Ca2+-ATPase.
Biophys. J., 2002, vol. 83, p.p. 2457-2474.
164. Liang, S., and Grishin, N. V., Effective scoring functions for protein sequence
design. Prot. Struct. Fun. Bioinfo., 2004, vol. 54, p.p. 271-281.

176
165. Liwo, A., et al., A United-Residue Force Field for Off-Lattice Protein-Structure
Simulations. I. Functional forms and parameters of long-range side-chain
interaction potentials from protein crystal data. J. Comput. Chem., 1997a, vol.
18, p.p. 849-873.
166. Liwo, A., et al., A United-Residue Force Field for Off-Lattice Protein-Structure
Simulations. II. Parameterization of short-range interactions and determination
of weights of energy terms by Z-score optimization. J. Comput. Chem., 1997b,
vol. 18, p.p. 874-887.
167. Looger, L. L., and Hellinga, H. W., Generalized dead-end elimination
algorithms make large-scale protein side-chain structure prediction tractable:
implications for protein design and structural genomics. JMB, 2001, vol. 307,
p.p. 429-445.
168. Looger, L. L., Dwyer, M. A., Smith, J. J., and Hellinga, H. W., Computational
design of receptor and sensor proteins with novel functions, Nature, 2003, vol.
423, p.p.185-190.
169. Luan, C. H., et al., Differential scaning calorimetry studies of NaCl effect on the
inverse temperature transition of some elastin-based polytetra-, polypenta-, and
polynanopeptides. Biopoly., 1991, vol. 31, p.p. 465-475.
170. Luenberger, D. G., Linear and non-linear programming; 2nd Ed. © Springer
Verlag.
171. Luthra, A., Jha, A. N., Ananthasuresh, G. K. and Vishveshwara, S., A Method
for Computing the Inter-Residue Interaction Potentials for reduced Amino Acid
Alphabet. J. Biosci., 2007, vol. 32 (5), p.p. 883-889.
172. Malakauskas, S. M., and Mayo, S. L., Design, structure and stability of a
hyperthermophilic protein variant. Nat. Struct. Biol., 1998, vol. 5, p.p. 470-475.
173. Mandell, D. J., and Kortemme, T., Backbone flexibility in computational protein
design. Curr. Op. Biotech., 2009, vol. 20, p.p. 420-428.
174. Mead, A., Review of the development of Multidimensional Scaling methods.
The Statistician, 1992, vol. 41 (1), p.p. 27-39.
175. Mendes, J., Guerois, R., and Serrano, L., Energy estimation in protein design.
Curr. Op. Struct. Biol., 2002, vol. 12, p.p. 441-446.

177
176. Metropolis, N., Rosenbluth, A. W., Rosenbluth, M. N., Teller, A. H., and Teller,
E., Equations of state calculations by fast computing machines. J. Chem. Phys.,
1953, vol. 21, p.p. 1087-1092.
177. Micheletti, C., et al., Elastic properties of proteins: insight on the folding process
and evolutionary selection of native structures. JMB, 2002, vol. 321, p.p. 909-
921.
178. Mirny, L., and Shakhnovich, E., How to derive a protein folding potential: A
new approach to an old problem. JMB, 1996, vol. 264, p.p. 1164-1179.
179. Mirny, L., and Shakhnovich, E., Universally conserved positions in protein
folds: reading evolutionary signals about stability, folding kinetics and function.
J. Mol. Biol, 1999, vol. 291, p.p. 177-196.
180. Mirny, L., and Shakhnovich, E., Evolutionary conservation of the folding
nucleus. J. Mol. Biol, 2001, vol. 308, p.p. 123-129.
181. Mirny, L., and Shakhnovich, E., Protein folding theory: from lattice to all atom
models. Ann. Rev. Biophys. Biomol. Struct., 2001, vol. 30, p.p. 361-396.
182. Miyazawa, S. and Jernigan. R. L., Estimation of effective interresidue contact
energies from protein crystal structures: quasi-chemical approximation.
Macromol., 1985, vol. 18, p.p. 534-552.
183. Miyazawa, S. and Jernigan, R. L., Residue-residue Potentials with a Favorable
Contact Pair term and an Unfavorable High Packing Density Term, for
Simulation and Threading. JMB, 1996, vol. 256, p.p. 623-644.
184. Miyashita, O., Wolynes, P. G., and Onuchic, J. N., Simple energy landscape
model for the kinetics of functional transitions in proteins. J. Phys. Chem. B.,
2005, vol. 109, p.p. 1959-1969.
185. Miyata, T., Miyazawa, S., and Yasunaga, T., Two types of amino acid
substitutions in protein evolution. J. Mol. Evol., 1979, vol. 12, p.p. 219-236.
186. Moffet, D. A., and Hecht, M. H., De novo proteins from combinatorial libraries.
Chem. Rev., 2001, vol. 101, p.p. 3191-3203.
187. Mondragon, A., Wolberger, C., and Harrison, S. C., Structure of phage 434 cro
protein at 2.35 angstroms resolution. JMB, 1989 b, vol. 205, p.p. 179-188.

178
188. Moser, R., et al., An artificial crystalline DDT-binding polypeptide. FEBS Lett.,
1983, vol. 157, p.p. 247-251.
189. Moult, J., Comparison of database potentials and molecular mechanics force
fields. Curr. Op. Struct. Biol., 1997, vol. 7, p.p. 194-199.
190. Morrissey, M. P., and Shakhnovich, E. I., Design of proteins with selected
thermal properties. Fold. Desg., 1996, vol. 1, p.p. 391-405.
191. Murphy, R. L., Wallqvist, A., and Levy, M. R., Simplified amino acid alphabets
for protein fold recognition and implications for folding. Prot. Engg., 2000, vol.
13, p.p. 149-152.
192. Nanias, M. , et al., Packing helices in proteins by global optimization of a
potential energy function. PNAS, 2003, vol. 100, no. 4, pp. 1706-1710.
193. Nelson, D. L., and Cox, M. M., Principles of Biochemistry, 5th Ed. © W. H.
Freeman and Company.
194. Offredi, F., et al., De novo backbone and sequence design of an idealized /α β -
barrel protein: evidence of stable tertiary structure. JMB, 2003, vol. 325, p.p.
163-174.
195. Oiki, S., et al., Bundles of amphiphatic transmembrane alpha-helices as a
structural motif for ion-conducting channel proteins: Studies on sodium
channels and acetylcholine receptors. Proteins, 1990, vol. 8, p.p. 226-236.
196. Onuchic, J. N., Luthey-Schulten, Z., and Wolynes, P. G., Theory of Protein
Folding: The Energy Landscape Perspective. Ann. Rev. Phys. Chem., 1997, vol.
48, p.p. 545-600.
197. Pande, V. S., Grosberg, A. Y., and Tanaka, T., Statistical mechanics of simple
models of protein folding and design. Biophys. J., 1997, vol. 73, p.p. 3192-3210.
198. Papalambros, P. P. and Wilde, D. J., Principles of optimal design – modeling
and computation. Cambridge University Press, 2000.
199. Pedersen, J. T., and Moult, J., Genetic algorithms for protein structure
prediction. Curr. Op. Struct. Biol., 1996, vol. 6, p.p. 227-231.
200. Pettersen et al., UCSF Chimera – a visualization system for exploratory research
analysis. J. Comput. Chem., 2004, vol. 25(13), p.p. 1605-1612.

179
201. Pierce, N. A., et al., Conformational splitting: a more powerful criterion for
dead-end elimination. J. Comput. Chem., 2000, vol. 21, p.p. 999-1009.
202. Plaxco, K. W., et al, Simplified proteins: minimalist solutions to the ‘protein
folding problem’. Curr. Op. Struct. Biol., 1998, vol. 8, p.p. 80-85.
203. Pokala, N., and Handel, T. M., Review: Protein design — Where We Were,
Where We Are, Where We’re Going. J. Struct. Biol., 2001, vol. 139, p.p. 269-
281.
204. Ponder, J. W., and Richards, F. M., Tertiary templates for proteins Use of
Packing criteria in the enumeration of allowed sequences for different structural
classes. JMB, 1987, vol. 193, p.p. 775-791.
205. Ptitsyn, O. B., and Ting, K. L. H., Non-functional conserved residues in globins
and their possible role as a folding nucleus. JMB, 1999, vol. 291, p.p. 671-682.
206. Quinn, T. P. et al, Betadoublet: De novo design, synthesis, and characterization
of a beta-sandwich protein. PNAS, 1994, vol. 91, p.p. 8487-8751.
207. Rader, A. J., and Bahar, I., Folding core predictions from network models of
proteins. Polym., 2004, vol. 45, p.p. 659-668.
208. Rader, A. J., et al., Identification of core amino acids stabilizing rhodopsin.
PNAS, 2004, vol. 101, p.p. 7246-7251.
209. Rader, A. J. et al., Maturation dynamics of HK97 bacteriophage capsid. Struct.,
2005, vol. 13, p.p. 413-421.
210. Raha, K., Wollacott, A. M., Italia, M. J., and Desjarlais, J. R., Prediction of
amino acid sequence from structure. Prot. Sci., 2000, vol. 9, p.p. 1106-1119.
211. Rakshit, S., and Ananthasuresh, G. K., An amino acid map of inter-residue
contact energies using metric multi-dimensional scaling. J. Theo. Biol., 2008,
vol. 250, p.p. 291-297.
212. Rakshit, S., and Ananthasuresh, G. K., A novel approach for large-scale
polypeptide folding based on elastic networks using continuous optimization. J.
Theo. Biol., 2010, vol. 262, p.p. 488-497.
213. Ramachandran, G. N., Ramakrishnan, C., Sasisekharan, V., Stereochemistry of
polypeptide chain configurations. JMB, 1963, vol. 7, p.p. 95-99.

180
214. Regan, L., and DeGrado, W. F., Characterization of Helical protein designed
from First Principles. Science, 1988, vol. 241, p.p. 976-978.
215. Rothlisberger, D., et al, Kemp elimination catalysis by computational enzyme
design. Nature, 2008, vol. 453, p.p. 190-195.
216. Roux, B., and Karplus, M., The normal modes of the Gramacidin-A dimmer
channel. Biophys. J., 1988, vol. 53, p.p. 297-309.
217. Sali, A., and Blundell, T. L., Comparative protein modeling by satisfaction of
spatial restraints. JMB, 1993, vol. 234, pp. 779-815.
218. Sancho, D. D., et al., Evolutionary Method for the Assembly of rigid protein
Elements. J. Comput. Chem., 2004, vol. 26, pp. 131-141.
219. Sander, C., Design of protein structures: helix bundles and beyond. Trends.
Biotech., 1994, vol. 12, p.p. 163-167.
220. Saven, J. G., and Wolynes, P. G., Statistical Mechanics of The Combinatorial
Synthesis and Analysis of Folding Macromolecules. J. Phys. Chem. B, 1997,
vol. 101, p.p. 8375-8389.
221. Saven, J. G., Combinatorial protein design. Curr. Op. Struct. Biol., 2002, vol.
12, p.p. 453-458.
222. Schueler-Furman, O., et al, Progress in Modeling of Protein Structures and
Interactions. Science, 2005, vol. 310, p.p. 638-642.
223. Schulz, G. E., and Schirmer, R. H., Principles of Protein Structure., Ed. Charles
R Cantor, (Springer advanced texts in chemistry) © Springer Verlag , 1979, pp.
10-16.
224. Schuyler, A. D., and Chirikjian, G. S., Normal mode analysis of proteins: a
comparison of rigid cluster modes with C-alpha coarse graining. J. Mol. Graph.
Model., 2004, vol. 22, p.p. 183-193.
225. Scrutton, N. S., et al., Redesign of the coenzyme specificity of a dehydrogenase
by protein engineering. Nature, vol. 343, p.p. 38-43.
226. Schwartz, R. M. and Dayhoff, M. O., Matrices for detecting distant
relationships. Atlas of Prot. Struct., 1978, vol. 5, p.p. 353-358.
227. Shakhnovich, E. I., and Gutin, A. M., Engineering of stable and fast-folding
sequences of model proteins. PNAS, 1993, vol. 90, p.p. 7195-7199.

181
228. Seno, F., et al., Optimal protein design procedure. PRL, 1996, vol. 77 (9), p.p.
1901-1904.
229. Shen, Y., et al., Intrinsic flexibility and gating mechanism of the potassium
channel KcsA. PNAS, 2002, vol. 99(4), p.p. 1949-1953.
230. Shewchuk, J. R., 1994, www.cs.cmu.edu/~quake-papers/painless-conjugate-
gradient.pdf (as on 28th December, 2009).
231. Shafmeister, C. E., et al, A designed four helix bundle protein with native-like
structure. Nat. Struct. Biol., 1997, vol. 4(12), p.p. 1039-1046.
232. Shafmeister, C. E., and Stroud, R. M., Helical protein design. Curr. Op.
Biotech., 1998, vol. 9, p.p. 350-353.
233. Shakhnovich, E. I., and Gutin, A. M., Engineering of stable and fast-folding
sequences of model proteins. PNAS, 1993, vol. 90, p.p. 7195-7199.
234. Siegel et al, Computational design of an enzyme catalyst for a stereoselective
biomolecular Diels-Alder reaction. Science, 2010, vol. 329, p.p. 309-314.
235. Simons, K. T., Kooperberg, C., Huang, E., and Baker, D., Assembly of protein
tertiary structures from fragments with similar local sequences using simulated
annealing and Bayesian scoring functions. JMB, 1997, vol. 268, p.p. 209-225.
236. Sinha, K. K. and Udgaonkar, J. B. Early events in protein folding. Curr. Sc.,
2009, vol. 96 (8), pp. 1053-1070.
237. Sippl, M. J., Calculation of conformational ensembles from potentials of mean
force. An approach to the knowledge-based prediction of local structures in
globular proteins. JMB, 1990, vol. 213, p.p. 859-883.
238. Sluka, J. P. et al., Synthesis of a sequence-specific DNA-cleaving peptide.
Science, 1987, vol. 238, p.p. 1129-1132.
239. Srivastava, I., and Bahar, I., Common mechanism of pore opening shared by
five different potassium channels. Biophys. J., 2006, vol. 90, p.p. 3929-3940.
240. Street, A. G., and Mayo, S. L., Computational protein design. Structure, 1999,
vol. 7(5), p.p. 105-109.
241. Su, A., and Mayo, S. L., Coupling backbone flexibility and amino acid sequence
selection in protein design. Prot. Sci., 1997, vol. 6, p.p. 1701-1707.

182
242. Svensson, L. A., Thulin, E., and Forsen, S., Proline cis-trans isomers in
calbindin D9K observed by X-ray crystallography. JMB, 1992, vol. 223, p.p.
601-606.
243. Sym, L., Taneja, A. K., and Hodges, R. S., Synthesis of a model protein of
defined secondary and quaternary structure. J. Bio. Chem., 1984, vol. 259, p.p.
13253-13261.
244. Szarecka, A., Xu, Y, and Tang, P., Dynamics of heteropentameric nicotinic
acetylcholine receptor: Implications of the gating mechanism. Proteins, 2007,
vol. 68, p.p. 948-960.
245. Taly, A., et al., Normal mode analysis suggests a quaternary twist model for the
nicotinic receptor gating mechanism, Biophys. J., 2005, vol. 88, p.p. 3954-3965.
246. Tama, F., et al., Building-block approach for determining low-frequency normal
modes of macromolecules. Proteins, 2000, vol. 41, p.p. 1-7.
247. Tama, F., et al., Exploring global distortions of biological macromolecules and
assemblies from low-resolution structural information and elastic network
theory. JMB, 2002, vol. 321, p.p. 297-305.
248. Tama, F., and Brooks III, C. L., Diversity and identity of mechanical properties
of icosahedral viral capsids studied with elastic network normal mode analysis.
JMB, 2005, vol. 345, p.p. 299-314.
249. Tanaka, S., and Scheraga, H. A., Medium- and long-range interaction
parameters between amino acids for predicting three-dimensional structures for
proteins. Macromol., 1976, vol. 9, p.p. 945-950.
250. Tatsumi, R., et al., A hybrid method of molecular dynamics and harmonic
dynamics for docking of flexible ligand to flexible receptor. J. Comput. Chem.,
2004, vol. 25, p.p. 1995-2005.
251. Tirion, M. M., Large amplitude elastic motions in proteins from a single-
parameter, atomic analysis. PRL, 1996, vol. 27(9), p.p. 1905-1908.
252. Thomas, P. D., and Dill, K. A., Statistical Potentials Extracted from Protein
Structures: How accurate are they ?. JMB, 1996, vol. 257, p.p. 457-469.
253. Torgerson, W. S., Multidimensional Scaling: I. Theory and Method.
Psychometrica, 1952, vol. 17 (4), p.p. 401-419.

183
254. Tozzini, V., Coarse-grained models for proteins. Curr. Op. Struct. Biol., 2005,
vol. 15, p.p. 144-150.
255. Tozzini, V., and Rocchia, W., Mapping All-Atom Models onto One-Bead
Coarse-Grained Models: General Properties and Applications to a Minimal
Polypeptide Model. J. Chem. Theory Comput., vol. 2, pp. 667-673, 2006.
256. Tozzini, V., Multiscale modeling of proteins. Acc. Chem. Res., 2010, vol. 43(2),
p.p. 220-230.
257. Tozzini, V., Minimalist models for proteins: a comparative analysis. Quat. Rev.
Biophy., 2010, vol. 43(3), p.p. 333-371.
258. Trylska, J., et al., Exploring global motions and correlations in the ribosome.
Biophys. J., 2005, vol. 89, p.p. 1455-1463.
259. Tuffery, P., et al, A new approach to the rapid determination of protein side
chain conformations. J. Biomol. Struct. Dyn, 1991, vol. 8, p.p. 1267-1269.
260. Valadie et al., Dynamical Properties of the MscL of Escherichia coli: A normal
mode analysis. JMB, 2003, vol. 332, p.p. 657-674.
261. Venkatarajan, M. S., and Braun, W., New quantitative descriptors of amino-
acids based on multidimensional scaling of a large number of physical-chemical
properties. J. Mol. Model, 2001, vol. 7, p.p. 445-453.
262. Ueda, Y., Taketomi, H., and Gō, N., Studies on protein folding, unfolding, and
fluctuations by computer simulation. II. A three-dimensional lattice model of
Lysozyme. Macromol., 1978, vol. 17, p.p. 1531-1548.
263. Urry, D. W., Protein Folding: Deciphering the Second Half of the Genetic Code.
American Assoc. for the Advancement of Science (Eds. L. Gierasch and J. King),
1990, p.p. 63-67.
264. Voigt, C. A., Gordon, D. B., and Mayo, S. L., Trading accuracy for speed: A
Quantitative Comparison of Search algorithms in Protein Sequence Design.
JMB, 2000, vol 299, p.p. 789-803.
265. Wächter, A., An Interior Point Algorithm for Large-Scale Nonlinear
Optimization with Applications in Process Engineering. PhD thesis, Carnegie
Mellon University, Jan. 29, 2002.

184
(http://www.research.ibm.com/people/a/andreasw/papers/thesis.pdf as on 24th
July, 2010).
266. Wächter, A., and Biegler, L. T., On the implementation of an interior-point filter
line-search algorithm for large-scale nonlinear programming. Mathematical
Programming, 2006, vol. 106 (1), p.p. 25-57.
267. Wang, J. and Wang, W., A computational approach to simplifying the protein
folding alphabet. Nat. Struct. Biol., 1999, vol. 6 (11), p.p. 1033-1038.
268. Wang, J., and Wang, W., Grouping of residue based on their contact
interactions. PRL E, 2002, vol. 65, doi. 041911-5.
269. Wernisch, L., Hery, S., and Wodak, S. J., Automatic protein design with all
atom force-fields by exact and heuristic optimization, 2000, vol. 301, p.p. 713-
736.
270. Wharton, R. P., and Ptashne, M., Changing the binding specificity of a represser
by redesigning an alpha-helix. Nature, 1985, vol. 316, p.p. 601-605.
271. Wolynes, P. G., As simple as can be ?, Nat. Struct. Biol., 1997, vol. 4 (11), p.p.
871-874.
272. Wolynes, P. G., Energy landscapes and solved protein-folding problems, Phil.
Trans. R. Soc. A., 2004, vol. 363, p.p. 453-467.
273. Woolfson, D. N., Core-directed protein design. Curr. Op. Struct. Biol., 2001,
vol. 11, p.p. 464-471.
274. Xia, Y., and Levitt, M., Simulating protein evolution in sequence and structure
space. Curr. Op. Struct. Biol., 2004, vol. 14, p.p. 2002-2007.
275. Xu, C., Tobi, D., and Bahar, I., Allosteric changes in protein structure computed
by a simple mechanical model: haemoglobin T↔R2 transition. JMB, 2003, vol.
333, p.p. 153-168.
276. Xu, J., and Zhang, Y., How significant is a protein structure similarity with TM-
score=0.5 ?. Struct. Bioinform., 2010, vol. 26 (7), p.p. 889-895.
277. Yan, Y., and Erickson, B. W., Engineering of betabellin 14D: Disulphide-
induced folding of a beta-sheet protein. Prot. Sci., 1994, vol. 3, p.p. 1069-1073.

185
278. Yang, L. W., and Bahar, I., Coupling between catalytic site and collective
dynamics: A requirement for mechanochemical activity of enzymes. Struct.,
2005, vol. 13, p.p. 893-904.
279. Yue, K., and Dill, K. A., Inverse protein folding problem: Designing polymer
sequences. PNAS, 1992, vol. 89, p.p. 4163-4167.
280. Yue, K., et al., A test of lattice protein folding algorithms. PNAS, 1995, vol. 92,
p.p. 325-329.
281. Yue, K., et al., Constraint-based assembly of tertiary protein structures from
secondary structure elements. Prot. Sci., 2008, vol. 9, pp. 1935-1946.
282. Zhang, L., and Skolnick, J., How do potentials derived from structural databases
relate to “true” potentials?. Prot. Sci., 1998, vol. 7, p.p. 112-122.
283. Zhang, Z., Shi, Y., and Liu, H., Molecular dynamics simulations of peptides and
proteins with amplified collective motions. Biophy. J., 2003, vol. 84, p.p. 3583-
3593.
284. Zhang, Y., and Skolnick, J., Scoring function for automated assessment of
protein structure template quality. Proteins, vol. 57, pp. 702-710, 2004.
285. Zhang, Y., and Skolnick, J., TM-align: a protein structure alignment algorithm
based on the TM-score. Nucleic Acids Res., vol. 33, no. 7, pp. 2302-2309, 2005.
286. Zou, J., and Saven, J. G., Statistical theory of combinatorial libraries of folding
polypeptides: energetic discrimination of a target structure. JMB, 2000, vol. 296,
p.p. 281-294.


















