49714248 USMLE Genetics Case Studies OSPE MCQs Dr Kumar Ponnusamy ST Matthew s University School of...
11
“Innovation requires energy. Energy comes from challenges that excite the imagination”
-
Upload
rilsac-cherian -
Category
Documents
-
view
122 -
download
4
description
x
Transcript of 49714248 USMLE Genetics Case Studies OSPE MCQs Dr Kumar Ponnusamy ST Matthew s University School of...

“Innovation requires energy.Energy comes from challenges that excite the imagination”

Down Syndrome: Trisomy 21 (47, XX, +21)
Signs & SymptomsSigns & Symptoms MCQMCQ
Answer: B. Answer: B. Aneuploidy: The category of chromosome changes which Aneuploidy: The category of chromosome changes which do not involve whole setsdo not involve whole sets. . It is usually the consequence of a It is usually the consequence of a failure of a single chromosome (or bivalent) to complete division.failure of a single chromosome (or bivalent) to complete division.
Y Chromosome is Missing
47, XX,+21 (Numerical Abnormality)Trisomy 21Trisomy 21
Most common autosomal aneuploid trisomy 21.Short stature, mental retardation, protruding tongue, Congenital heart defects, kidney defects, suppressed immune systems, digestive disorders. Depressed nasal bridge, Upslanting palpebral fissures, Epicanthal fold.
Down syndrome, trisomy 21Down syndrome, trisomy 21The incidence of trisomy 21 rises sharply with The incidence of trisomy 21 rises sharply with increasing maternal age.increasing maternal age.Most cases arise from Most cases arise from non disjunction in the non disjunction in the first meiotic divisionfirst meiotic division, the father contributing the extra , the father contributing the extra chromosome in 15% of cases.chromosome in 15% of cases.
A small proportion of cases are A small proportion of cases are mosaicmosaic and these probably arise from a non disjunction event in and these probably arise from a non disjunction event in an early zygotic division.an early zygotic division.
About About 4% of cases4% of cases arise by inheritance of a arise by inheritance of a translocation chromosometranslocation chromosome from a parent who is a from a parent who is a balanced carrier. The symptoms include characteristic balanced carrier. The symptoms include characteristic facial dysmorphologies, and an IQ of less facial dysmorphologies, and an IQ of less than 50than 50. Down syndrome is responsible for about 1/3 of all cases of moderate to . Down syndrome is responsible for about 1/3 of all cases of moderate to severe mental severe mental handicaphandicap..
A newborn infant with a A newborn infant with a single palmar creasesingle palmar creaseand and depressed nasal bridgedepressed nasal bridge is suspected to is suspected to have a genetic disorder. The karyotype have a genetic disorder. The karyotype analysis shows the presence of 47, XX,+21. analysis shows the presence of 47, XX,+21. The abnormality that results in this The abnormality that results in this karyotype is:karyotype is:
A. Nondisjunction during spermatogenesis.A. Nondisjunction during spermatogenesis.B. Nondisjunction during oogenesis.√B. Nondisjunction during oogenesis.√C. Presence of an extra X chromosome.C. Presence of an extra X chromosome.D. Faulty X-inactivation during bryogenesis.D. Faulty X-inactivation during bryogenesis.

Retinoblastoma: Autosomal dominant Retinoblastoma: Autosomal dominant
RetinoblastomaRetinoblastoma
Retinoblastoma is caused by an inherited loss-of-Retinoblastoma is caused by an inherited loss-of-function mutation in the Rb tumor suppressor function mutation in the Rb tumor suppressor gene. In 10% of individuals who inherit this gene. In 10% of individuals who inherit this mutation, there is no additional somatic mutation in mutation, there is no additional somatic mutation in the normal copy ad retinoblastoma does not the normal copy ad retinoblastoma does not develop, although they can pass the muttion to develop, although they can pass the muttion to their offspring. Penetrance of rtinoblastoma is their offspring. Penetrance of rtinoblastoma is therefore 90%. therefore 90%.
Answer: C. Answer: C. Because multiple family members are affected and because mutations at the Because multiple family members are affected and because mutations at the retinoblastoma gene are known to be sometimes nonpenetrant, the man in question is most retinoblastoma gene are known to be sometimes nonpenetrant, the man in question is most likely an obligate carrier of the mutation who did not experience a second mutation in this likely an obligate carrier of the mutation who did not experience a second mutation in this gene during his fetal development. gene during his fetal development.
A 20 year-old man has had no retinoblastomas A 20 year-old man has had no retinoblastomas but has produced 2 offspring with multiple but has produced 2 offspring with multiple retinoblastomas. In addition, his father had two retinoblastomas. In addition, his father had two retinoblastomas as a young child, and one of his retinoblastomas as a young child, and one of his siblings had 3 retinoblsatomas. What is the most siblings had 3 retinoblsatomas. What is the most likely explanation for the absence of likely explanation for the absence of retinoblastomas in this individual?retinoblastomas in this individual?
A. A new mutation in the unaffecteed individual, A. A new mutation in the unaffecteed individual, which has corrected the disease causing which has corrected the disease causing mutation.mutation.B. Highly variable expression of the disorder.B. Highly variable expression of the disorder.C. Incomplete penetrance.√C. Incomplete penetrance.√D. Multiple new mutations in other family D. Multiple new mutations in other family members.members. E. Pleiotrpy.E. Pleiotrpy.
Incomplete penetrance in Familial Cancer / Double Hit HypothesisIncomplete penetrance in Familial Cancer / Double Hit Hypothesis
Incomplete penetrance: Incomplete penetrance: Referring to the presence of a gene that is not phenotypically expressed in all members of a family with the gene.
In non-inherited retinoblastoma, two "hits" had to take place before a tumor could develop, explaining the age difference. It was later found that carcinogenesis (the development of malignancy) depended both on the activation of proto-oncogenes (genes that stimulate cell proliferation) and deactivation of tumor suppressor gene (TSG) (genes that keep proliferation in check). A first "hit" in an oncogene would not necessarily lead to cancer, as normally functioning TSGs would still counterbalance this impetus; only damage to TSGs would lead to unchecked proliferation. Conversely, a damaged TSG (such as the Rb1 gene in retinoblastoma) would not lead to cancer unless there is a growth impetus from an activated oncogene.
The Knudson hypothesis is the hypothesis that cancer is the result of accumulated mutations to a cell's DNA. Knudson performed a statistical analysis on cases of retinoblastoma, a tumor of the retina which occurs both as an inherited disease and sporadically. Inherited retinoblastoma occurs at a younger age than the sporadic disease. In addition, the children with inherited retinoblastoma often developed the tumor in both eyes, suggesting an underlying predisposition. Knudson suggested that multiple "hits" to DNA were necessary to cause cancer. In the children with inherited retinoblastoma, the first insult was inherited in the DNA, and any 2nd insult would rapidly lead to cancer.
Knudson’s Double-Hit HypothesisKnudson’s Double-Hit Hypothesis

Angelman / Happy Puppet Syndrome: Imprinting, Affects ♂ & ♀
Case ScenarioCase Scenario
Signs & SymptomsSigns & Symptoms
MCQMCQ
Answer: A. Answer: A. Imprinting refers to the differential transcriptional activity of genes inherited from the Imprinting refers to the differential transcriptional activity of genes inherited from the father verses the mother.father verses the mother.
Location: Chromosome 15.
Happy disposition, severe mental retardation, seizures., ataxia, puppet-like posture of limbs, "Happy Puppet syndrome“. Puppet like posture of limbs.
Problem: Deletion of maternal 15q11-13.
Angelman syndrome is produced if there is a deletion of 15q11-13 from the maternal chromosome. This has led to the conclusion that thee are atleast two imprinted genes within this region, one active on the paternal chromosome 15 & the other normally active on the maternal chromosome 15. Loss, usually by deletion of paternal 15q11-13, causes Prader-Willi, whereas loss of the maternal 15q11-13 causes Angelman syndrome ..
In studying a large number of families with a In studying a large number of families with a small deletion in a specific chromosome region, small deletion in a specific chromosome region, it is noted that the disease phenotype is distinctly it is noted that the disease phenotype is distinctly different when the deletion is inherited from the different when the deletion is inherited from the mother as opposed to the father. What is the mother as opposed to the father. What is the most likely explanation. most likely explanation.
A.A.Imprinting.√Imprinting.√B.B.Mitochondrial inheritance.Mitochondrial inheritance.C.C.Sex-dependent penetrance.Sex-dependent penetrance.D.D.X-linked dominant inheritance.X-linked dominant inheritance.E.E.X-linked recessive inheritance.X-linked recessive inheritance.
Imprinting Imprinting refers to the fact that a small number of genes are transcriptinally active only when refers to the fact that a small number of genes are transcriptinally active only when transmitted by one of the two sexes. The homologous locus in the other parent is rendered transmitted by one of the two sexes. The homologous locus in the other parent is rendered transcriptionally inactive. Thus, for imprinted loci, it is normal to have only the maternal (for some transcriptionally inactive. Thus, for imprinted loci, it is normal to have only the maternal (for some loci) active, or only the paternal (for other loci) active.loci) active, or only the paternal (for other loci) active.
Imprinting: Imprinting: occurs during gametogenesis.occurs during gametogenesis.Is maintained in all somatic cells of the offspring.Is maintained in all somatic cells of the offspring.During gametogenesis in the offspring, is erased & re-established according to the sex of the During gametogenesis in the offspring, is erased & re-established according to the sex of the individual.individual.Involves methylation & possibly other mechanisms to imprint or inactivate the appropriate loci.Involves methylation & possibly other mechanisms to imprint or inactivate the appropriate loci.Occurs in specific loci on several chromosomes. Occurs in specific loci on several chromosomes.
ImprintingImprintingThe process by which maternally and paternally derived chromosomes are uniquely chemically modified leading to different expression of a certain gene or genes on those chromosomes depending on their parental origin.
A phenomenon in which the disease phenotype depends on which parent passed on the disease gene. For instance, both Prader-Willi and Angelman syndromes are inherited when the same part of chromosome 15 is missing. When the father's complement of 15 is missing, the child has Prader-Willi, but when the mother's complement of 15 is missing, the child
has Angelman syndrome.

Turners Syndrome (TS) ♀ (45, X or 45, X0)
45 X0, Note the unpaired X at lower right, at 2345 X0, Note the unpaired X at lower right, at 23rdrd pair. pair.
Karyotype ResultsKaryotype Results
Turner Syndrome (45, X or 45, X0)Turner Syndrome (45, X or 45, X0)An 18 year old female has presented to the An 18 year old female has presented to the physician with primary amenorrhea. On physician with primary amenorrhea. On examination she has a characteristic webbing examination she has a characteristic webbing of the neck. Karyotype analysis of the subject of the neck. Karyotype analysis of the subject would most likely reveal,would most likely reveal, A. 47, XX,+21.A. 47, XX,+21.B. 46, XX.B. 46, XX.C. 47, XXY.C. 47, XXY.D. 47, XYY.D. 47, XYY.E. 45, X0. √E. 45, X0. √
Answer: E. Answer: E.
Problem: Problem: Many are mosaic. Some of their Many are mosaic. Some of their cells are 45, X0. Other cells are 46 XX & 47, cells are 45, X0. Other cells are 46 XX & 47, XXX. Cells with 45, X; have no Barr body. XXX. Cells with 45, X; have no Barr body.
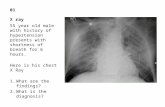
Only monosomy consistent with life.Only monosomy consistent with life.50% are 45, X.50% are 45, X.Majority of others are mosaics for 45, X & Majority of others are mosaics for 45, X & one other cell lineage (46, XX, 47,XXX, ect.,).one other cell lineage (46, XX, 47,XXX, ect.,).Short stature. Short stature. Edema of wrist & ankles in newborn.Edema of wrist & ankles in newborn.Cystic hygroma in utero resulting in excess Cystic hygroma in utero resulting in excess nuchal skin & “webbed” neck.nuchal skin & “webbed” neck.Primary amenorhea.Primary amenorhea.Coarctation of the aorta or other congenital Coarctation of the aorta or other congenital heart defect in some cases.heart defect in some cases.Infertility.Infertility.Gonadal dysgenesis.Gonadal dysgenesis.
Genetic Mosaicism in Turners SyndromeGenetic Mosaicism in Turners Syndrome
Genetic mosaicism is defined as a condition in which there are cells of different genotypes Genetic mosaicism is defined as a condition in which there are cells of different genotypes or chromosome constitutions within a single individual. Some women with Turner or chromosome constitutions within a single individual. Some women with Turner syndrome have somatic cells that are 45, x and others that are 46, XX or 47,XXX. Mosaicism syndrome have somatic cells that are 45, x and others that are 46, XX or 47,XXX. Mosaicism in Turner syndrome is thought to arise in early embryogenesis by mechanism that are not in Turner syndrome is thought to arise in early embryogenesis by mechanism that are not completely understood. completely understood.

Prader Willi Syndrome (PWS) – Imprinting, Affects ♂ & ♀
Case ScenarioCase Scenario
Signs & SymptomsSigns & Symptoms
MCQMCQ
Answer: D. Answer: D. These syndromes involve reciprocal imprinting of one or more genes in a region of the long These syndromes involve reciprocal imprinting of one or more genes in a region of the long arm of chromosome 15. Although both diseases manifest neurologic impairment, Angelman syndrome is by arm of chromosome 15. Although both diseases manifest neurologic impairment, Angelman syndrome is by far the more severe of the two disorders. far the more severe of the two disorders.
Neonatal hypotonia, poor feeding in neonatal period, behavior problems, moderate mental & developmental problems, developmental delay, hypogonadism & underdeveloped genitalia, hypotonia in infancy, failure to thrive, hyperphgia / overeating & obesity by age 2-4 yrs, small hands & feet, very low recurrence risk.
Chromosome 15
Deletion of 15q11-13
Paternal Maternal
p arm
q arm
A 3-year-old boy is evaluated for obesity. At birth A 3-year-old boy is evaluated for obesity. At birth he fed poorly and was some what hypotonic and he fed poorly and was some what hypotonic and lethargic. At the time he was diagnosed with lethargic. At the time he was diagnosed with failure to thrive, cause unknow, and was given failure to thrive, cause unknow, and was given intragastric feeding s until he ragained his birth intragastric feeding s until he ragained his birth weight. He continued to gain weight slowly but weight. He continued to gain weight slowly but remained in the lowest quadrile for age-remained in the lowest quadrile for age-appropriate weight and height. Walking was appropriate weight and height. Walking was delayed until he was 26 month old. Over the last delayed until he was 26 month old. Over the last year his appetite was increased dramatically. He year his appetite was increased dramatically. He was begun having temper tantrums of increasing was begun having temper tantrums of increasing frequency and violence., causing his withdrawal frequency and violence., causing his withdrawal from preschool. His current evaluation reveals an from preschool. His current evaluation reveals an obese boy with mental and developmental delay. obese boy with mental and developmental delay. The physician also notes underdeveloped The physician also notes underdeveloped genitalia, and she refers the boy to genetics clinic genitalia, and she refers the boy to genetics clinic for karyotype analysis. The result shows a for karyotype analysis. The result shows a deletion from one copy of cheromosome 15q11-deletion from one copy of cheromosome 15q11-q13 consistent with Prader-Willi Syndrome. q13 consistent with Prader-Willi Syndrome.
Problem:Deletion of paternal 15q11-13
Location: Chromosome 15.
Angelman and Prader-Willi syndromes are related disorders affecting genes on chromosome 15 by which of the following epigenetic mechanisms?.A.Mosaicism.B.Histone acetylation.C.Haploinsufficiency.D.Imprinting.√E.Viral infection.
Imprinting: In genomic imprinting the ability of a gene to be expressed depends upon the sex of the parent who passed on the gene. In some cases imprinted genes are expressed when the are inherited from the mother. in other cases they are expressed when inherited from the father. imprinting affects gene expression by chemically modifying DNA and/or altering the chromatin structure.
E.g. PWS & Angelman syndrome (AS) are two distinct diseases caused by a deletion in the same part of chromosome 15. When this deletion occurs on the chromosome 15 that came from the father, the child will have PWS. However, when the deletion occurs on the chromosome 15 that came from the mother, the child will develop AS.

Marfan Syndrome: Autosomal Dominant, Pleiotropy Marfan Syndrome: Autosomal Dominant, Pleiotropy
Pleotropy: Pleotropy: Pleiotropy occurs when a single gene influences multiple pheotypic traits. Consequently, a new mutation in the gene may have an effect on some or all traits simultaneously. This can become a problem when selection on one trait favors one specific version of the gene (allele), while the selection on the other trait favors another allele. Mutation Mutation in this type of gene will simultaneously affect more than one trait. An example is phenylketonuria, a human disease caused by muttion(s) in a single gene that codes for the enzyme , phenylalanine hydroxylase. The disease is characterized by mental retardation and reduced hair and skin pigmentation.
A disease causing mutation affects multiple A disease causing mutation affects multiple organ systems. Mutation in the fibrillin gene.organ systems. Mutation in the fibrillin gene.Skeletal abnormalities (long limbs, pectus Skeletal abnormalities (long limbs, pectus excavatum). Hyper-mobile joints. Ocular excavatum). Hyper-mobile joints. Ocular abnormalities (myopia, lens dislocation).abnormalities (myopia, lens dislocation).Cardiovascular disease (Mitral valve prolapse, Cardiovascular disease (Mitral valve prolapse, Aortic aneuysm). Oseogeness imperfects Aortic aneuysm). Oseogeness imperfects (Bone, sclera). (Bone, sclera).
Signs & SymptomsSigns & Symptoms MCQMCQ
Answer: D. Answer: D. For an autosomal dominant condition, the first occurrence in a family is usually the For an autosomal dominant condition, the first occurrence in a family is usually the result of a new mutation that occurred in one of the gametes transmitted by a parent of the result of a new mutation that occurred in one of the gametes transmitted by a parent of the affected individual.affected individual.
A 10-year-old girl is diagnosed with Marfan A 10-year-old girl is diagnosed with Marfan syndrome, an autosomal dominant condition.syndrome, an autosomal dominant condition.An extensive review of her pedigree indicaes An extensive review of her pedigree indicaes no previous family history of this disorder. The no previous family history of this disorder. The most likely explanation for this pattern is:most likely explanation for this pattern is:
A.A.highly variable expression of the disease highly variable expression of the disease phenotype.phenotype.B.B.incomplete penetrance. incomplete penetrance. C.C.mitochondrial compensation in the mother.mitochondrial compensation in the mother.D.D.new mutation transmitted by one of the new mutation transmitted by one of the parents to the affected girl.parents to the affected girl.E.E.pleiotrophy.pleiotrophy.
MarfanMarfan SyndromeSyndrome
Ocular abnormalities Ocular abnormalities (myopia, lens dislocation(myopia, lens dislocation
Skeletal abnormalities Skeletal abnormalities (long limbs, (long limbs, pectuspectus
excavatumexcavatum).).HyperHyper--mobile jointsmobile joints
Pleotropy
Pleiotropic:Pleiotropic: 1. Producing many effects. 2. 1. Producing many effects. 2. Multiple Multiple effects from a single geneeffects from a single gene.. For example, the For example, the Marfan gene is pleiotropicMarfan gene is pleiotropic with widespread effects with widespread effects and can cause long fingers and toes and can cause long fingers and toes ((arachnodactylyarachnodactyly), dislocation of the ), dislocation of the lens lens of the of the eyeeye, , and and dissecting aneurysmdissecting aneurysm of the of the aortaaorta. .

Gout Syndrome: Gout Syndrome: Degradation of Purine Nucleotides to Uric AcidDegradation of Purine Nucleotides to Uric Acid
Ans: B. Ans: B. This patient shows many symptoms consistent This patient shows many symptoms consistent with an episode of gout. His joint pain is due t gouty with an episode of gout. His joint pain is due t gouty arthritis, an infammatory condition arising from deposition arthritis, an infammatory condition arising from deposition of urate crystals. He swelling in the joints of his big toe of urate crystals. He swelling in the joints of his big toe (tophaceous gout) is also a manifestation of this (tophaceous gout) is also a manifestation of this phenomenon. In his case, the episode seems to have phenomenon. In his case, the episode seems to have been triggered by excessive eating at Thanksgiving been triggered by excessive eating at Thanksgiving dinner along with alcohol consumption, leading to dinner along with alcohol consumption, leading to degradation of large quantities of purine nucleotides and degradation of large quantities of purine nucleotides and consequent increased flux through the pathway that consequent increased flux through the pathway that produces uric acid. produces uric acid.
Gout: Gout: Recurrent acute arthritis Recurrent acute arthritis of of peripheral joints caused by the accumulation of peripheral joints caused by the accumulation of monosodium urate crystalsmonosodium urate crystals. Often presents as . Often presents as pain and swelling confined to one joint. The big pain and swelling confined to one joint. The big toe joint is commonly affected. The problems toe joint is commonly affected. The problems partly arise because partly arise because neutrophils release neutrophils release lysosomal enzymeslysosomal enzymes as a result of damage to as a result of damage to the phagosome membrane by ingested the phagosome membrane by ingested crystals: crystals: Colchicine Colchicine acts to reduce the attack by acts to reduce the attack by
inhibiting lysosome phagosome fusion.inhibiting lysosome phagosome fusion. . .
Signs & SymptomsSigns & Symptoms MCQMCQ
Answer: B. Answer: B. Whether his gout arises from impaired excretion of uric acid or is due to a mutation of Whether his gout arises from impaired excretion of uric acid or is due to a mutation of PRPP synthase cannot be determined from the data. Analysis of his blood may confirm the gout if PRPP synthase cannot be determined from the data. Analysis of his blood may confirm the gout if high concentrations of uric acid (hyperurecemia) are present.high concentrations of uric acid (hyperurecemia) are present. ..
A 47-year-old man complains of pain in the A 47-year-old man complains of pain in the joints of his big toe, which are obviously joints of his big toe, which are obviously swollen and tender. The pain has been chronic swollen and tender. The pain has been chronic but became intolerable the day after but became intolerable the day after thanksgiving when he had a large meal and thanksgiving when he had a large meal and several glasses of red wine. He is obese, andseveral glasses of red wine. He is obese, andHis past medical history is significant for His past medical history is significant for removal of kidney stones. removal of kidney stones. Which of the following Which of the following involved in the pathophysiology of this patient’s involved in the pathophysiology of this patient’s condition?.condition?.A.A. Elevated orotic acid.Elevated orotic acid.B.B. Elevated uric acid.Elevated uric acid.C.C. Deficiency of folic acid.Deficiency of folic acid.D.D. Anemia.Anemia.E.E. Hypoglycemia Highly Hypoglycemia Highly
Tophaeceous GoutTophaeceous GoutMonosodium Monosodium Urate CrystalsUrate Crystals

Autosomal Dominant Inheritance
Disease Repeat Description
Myotonic dystrophy
CTG Facial/extrremity weakness, cardiomyopathy, GI tract obstruction, respiratory tract muscle eakness, cataracts, testicular atrophy & mental retardation.
Fragile X syndrome
CGG Mental retardation, macro-orchism in men, large ears, prominent jaw
Friedreich’s ataxia
GAA Cerebellar dysfunction, limb ataxia, sensory defects, cardiomyopathy
Huntington’s disease
CAG Loss of motor control (chorea), dementia
Spinal & bulbar muscular atrophy
CAG Lower motor neuron disease, androgen insensitivity
Diseases Associated With Trinucleotide Repeat ExpansionDiseases Associated With Trinucleotide Repeat Expansion

X-Linked Dominant Inheritance
Disease Inheritance pattern
Anticipation
Parental sex bias for
instability
Repeat
Fragile X X-linked dominant
partial penetrance in
females
Yes Large expansion
only maternal origin
CGG
Spinal & bulbar muscular atrophy
X-linked recessive
No Predominantly parental
CAG
Myotonic dystrophy
AD Yes No CTG
Huntington’s disease
AD Yes, but rare
Parental CAG
Spinocerebellar ataxia 1
AD Yes Unknown CAG
Triple Repeat SequenceTriple Repeat Sequence

Variable Penetrance
Genetic principle Definition Examples
Variable expression
Variation in the severity of the disease due to environmental
effects, allelic heterogeneity, or interaction with other (modifier)
gene
NF1, osteogenesis
imperfecta (OI)
Allelic heterogeneity
Variable expression of a disease phenotype based on type of mutation at a disease locus
Osteogenesis imperfecta
(OI)
Reduced penetrance
When having a disease genotype does not necessarily produce the
disease phenotype
Retinoblastoma
(Rb)
Pleiotrophy Ability of a gene to cause many pehotypes
NF1, Marfan’s syndrome (MS), CF
Locus heterogeneity
When mutations at different gene loci can cause the same disease
phenotype
Adult Polycystic
Kidney Disease (APKD)