
2.6.1 緒言...ナルフラフィン塩酸塩 2.6.1 緒言 Page 5...
127
ナルフラフィン塩酸塩 医薬品製造販売承認申請書添付資料 2.6.1 緒言 東レ・メディカル株式会社
Transcript of 2.6.1 緒言...ナルフラフィン塩酸塩 2.6.1 緒言 Page 5...

ナルフラフィン塩酸塩
医薬品製造販売承認申請書添付資料
2.6.1 緒言
東レ・メディカル株式会社

ナルフラフィン塩酸塩 2.6.1 緒言 Page 2
目次
2.6.1 緒言 ......................................................................................................................................... 4
- 2 -

ナルフラフィン塩酸塩 2.6.1 緒言 Page 3
用語および略語一覧
用語および略語 説明 nor-BNI ノルビナルトルフィミン 2 塩酸塩 1 水和物(オピオイド κ 受容体拮抗薬、
Nor-binaltorphimine dihydrochloride monohydrate) PBC 原発性胆汁性肝硬変(Primary Biliary Cirrhosis) TRK-820 ナルフラフィン塩酸塩 β-エンドルフィン オピオイド μ 受容体作動性を有する内因性オピオイドペプチド Child-Pugh 分類 肝疾患の重症度分類 Leu-エンケファリン オピオイド μ 受容体作動性を有する内因性オピオイドペプチド Met-エンケファリン オピオイド μ 受容体作動性を有する内因性オピオイドペプチド エンドモルフィン-1 オピオイド μ 受容体作動性を有する内因性オピオイドペプチド サブスタンス P サブスタンス P 酢酸塩水和物(起痒剤) デオキシコール酸 デオキシコール酸ナトリウム 1 水和物(起痒剤) ナルトレキソン ナルトレキソン塩酸塩(オピオイド μ 受容体拮抗薬) ナロキソン ナロキソン塩酸塩(オピオイド μ 受容体拮抗薬) ヒスタミン ヒスタミン(起痒剤) モルヒネ モルヒネ塩酸塩(麻薬性鎮痛薬)
- 3 -

ナルフラフィン塩酸塩 2.6.1 緒言 Page 4
2.6.1 緒言
TRK-820 は、東レ株式会社基礎研究所(現 医薬研究所)において創製され、難治性のそう痒
症の治療を目的として開発されたモルヒナン骨格を有する止痒薬である(図 2.6.1-1)。TRK-820
は、先に血液透析患者における既存治療抵抗性のそう痒症を対象に臨床開発が行われ、国内では
東レ株式会社が経口剤として、2009 年 1 月に製造販売承認を取得し、同年 3 月よりレミッチ®カ
プセル 2.5 μg として販売が開始されている。今回、TRK-820 の経口剤について慢性肝疾患患者に
おける難治性そう痒症を対象として臨床開発を実施し、ノピコール®カプセル 2.5 μg として製造販
売承認申請を行うこととした。
(2E)-N-[(5R,6R)-17-(Cyclopropylmethyl)-4,5-epoxy-3,14-dihydroxymorphinan-6-yl]-3-(furan-3-yl)-N-
methylprop-2-enamide monohydrochloride
図 2.6.1-1. TRK-820 の構造式および化学名
一般的に痒みは、ヒスタミン、サブスタンス P、トリプターゼなどのケミカルメディエーター
による刺激が一次感覚神経を介して脊髄後角に入力した後、二次神経である脊髄視床路を通って
上行し 1)、視床に入力した後、大脳皮質にある一次および二次体性感覚野、帯状回皮質、島など
に伝達されると考えられている 2), 3)。さらに、中枢神経系において、痒みがオピオイド μ 受容体
の活性化を介して発現している可能性が示されている 3)。
一方、肝炎、肝硬変、閉塞性黄疸などの慢性肝疾患患者では、しばしば胆汁の流出が停滞する
「胆汁うっ滞」を呈し、全身性の強い痒みが生じることがある 4)-6)。また、特定疾患に指定されて
いる PBC はそう痒を主訴とし、患者の活動性や睡眠を著しく阻害することもある 7)。これらの慢
性肝疾患におけるそう痒症には複数の因子が関与していると考えられており、抗ヒスタミン薬な
どの既存治療薬に抵抗性であることが多いといわれている 8),9)。
慢性肝疾患患者では、血漿中の Leu-エンケファリンおよび Met-エンケファリン(オピオイド μ
受容体作動性を有する内因性オピオイドペプチド)濃度が高いことが報告されている 10),11)。また、
痒みを有する PBC 患者では、痒みを有さない PBC 患者と比較して、血漿中の β-エンドルフィン
およびエンドモルフィン-1(オピオイド μ 受容体作動性を有する内因性オピオイドペプチド)濃
. HCl
HO
H
HO
N
O H HCH3
O
O
N
- 4 -

ナルフラフィン塩酸塩 2.6.1 緒言 Page 5
度が高いことも報告されている 12)。さらに、胆汁うっ滞患者のそう痒に対してオピオイド μ 受容
体拮抗薬のナロキソン 13)もしくはナルトレキソン 14),15)が有効であるとの報告もある。したがって、
肝疾患患者の痒みの発現には、オピオイド μ 受容体の活性化が関与していることが示唆されてい
る。
また、オピオイド受容体のサブタイプ(μ、κ および δ)のうち、オピオイド κ 受容体は、一般
的にオピオイド μ 受容体と相反する作用を有することが知られている 16)。したがって、オピオイ
ド κ 受容体作動薬が、慢性肝疾患患者における難治性のそう痒症などのオピオイド μ 受容体の活
性化が関与すると考えられている痒みに対して有効である可能性が考えられた。
このような背景の下、オピオイド κ 受容体作動薬として見出された TRK-820 の難治性のそう痒
症に対する研究を実施し、そう痒症モデルにおける、TRK-820 の有効性が確認されている 17)。
今回の申請にあたり、本薬の薬理学的、薬物動態学的および毒性学的特徴を明らかにする目的
で各種試験を実施した。以下に TRK-820 の薬理学的特徴について簡潔に示した。
ヒトオピオイド受容体発現細胞を用いた in vitroの受容体結合試験および受容体作動性試験の結
果から、TRK-820 は選択的なオピオイド κ 受容体作動薬であることが示された。また、in vitro に
おいて、TRK-820 はヒスタミン受容体を含むオピオイド受容体以外の種々の受容体、イオンチャ
ネルおよびトランスポーターに結合せず、肥満細胞からの脱顆粒反応に対しても抑制作用を示さ
なかった。さらに、in vivo において、TRK-820 の経口投与によるサブスタンス P 皮内投与誘発マ
ウス引っ掻き行動抑制作用は、オピオイド κ 受容体拮抗薬である nor-BNI の脳室内投与により完
全に拮抗された。以上のことから、TRK-820 は、中枢神経系のオピオイド κ 受容体の活性化を介
して止痒作用を示すものと考えられた。
慢性肝疾患患者のそう痒発現には複数の因子が関与していると考えられていること、さらに、
この痒みが既存治療薬抵抗性であることが多いといわれていることから、既存の止痒薬である抗
ヒスタミン薬に対して感受性および抵抗性の各種そう痒モデルに対するTRK-820の作用を網羅的
に検討し、慢性肝疾患患者の痒みに対する有効性について考察した。
抗ヒスタミン薬が有効なヒスタミン皮内投与誘発マウス引っ掻き行動および抗ヒスタミン薬が
十分な効果を示さないサブスタンス P 皮内投与誘発マウス引っ掻き行動を指標にしたそう痒モデ
ルおいて、TRK-820 は経口投与によって引っ掻き行動を抑制したことから、止痒作用を有するこ
とが示唆された。また、抗ヒスタミン薬が無効な胆汁うっ滞性のそう痒モデルであるデオキシコ
ール酸皮内投与誘発マウス引っ掻き行動および NC/Nga 系マウスを用いた自然発症アトピー性皮
膚炎モデルの引っ掻き行動に対しても、TRK-820 は経口投与により引っ掻き行動を抑制した。さ
らに、抗ヒスタミン薬が無効な中枢性のそう痒モデルであるモルヒネ大槽内投与誘発マウス引っ
掻き行動に対して、TRK-820 は皮下投与により引っ掻き行動の抑制作用を示した。以上のように、
既存治療薬感受性および抵抗性の各種そう痒モデルすべてにおいて止痒作用の指標となる引っ掻
- 5 -

ナルフラフィン塩酸塩 2.6.1 緒言 Page 6
き行動抑制作用が認められたことから、TRK-820 は慢性肝疾患患者における難治性そう痒症に対
する止痒薬として有効性が十分期待できるものと考えられた。
申請した「効能又は効果」および「用法及び用量」を以下に記載した(表 2.6.1-1)。
表 2.6.1-1. TRK-820 の「効能又は効果」および「用法及び用量」(案)
効能又は効果 慢性肝疾患患者におけるそう痒症の改善(既存治療で効果不十分な場合に限る)
用法及び用量
通常、成人には、ナルフラフィン塩酸塩として 1 日 1 回 2.5 μg を夕食後又は就
寝前に経口投与する。なお、症状に応じて増量することができるが、1 日 1 回 5 μgを限度とする。 【用法及び用量に関連する使用上の注意】 本剤の投与は 1 日 1 回 2.5 μg から開始し、効果不十分な場合に 1 日 1 回 5 μg へ
の増量を検討すること。
- 6 -

ナルフラフィン塩酸塩 2.6.1 緒言 Page 7
参考文献
1) Andrew D, Craig AD. Spinothalamic lamina I neurons selectively sensitive to histamine: a central neural pathway for itch. Nat Neurosci. 2001;4:72-7. 【4.3-44】
2) Mochizuki H, Tashiro M, Kano M, Sakurada Y, Itoh M, Yanai K. Imaging of central itch modulation in the human brain using positron emission tomography. Pain. 2003;105:339-46. 【4.3-38】
3) Paus R, Schmelz M, Bíró T, Steinhoff M. Frontiers in pruritus research: scratching the brain for more effective itch therapy. J Clin Invest. 2006;116:1174-85. 【4.3-53】
4) 東田千尋. 肝障害のかゆみとオピオイド. 医学のあゆみ. 2001;197:616-7. 【4.3-10】
5) 伊崎誠一. Q30 肝疾患に伴うかゆみについて. In:宮地良樹編. かゆみ Q&A. 医薬ジャーナル
社, 1997;76-7. 【4.3-11】
6) Jones EA, Bergasa NV. The pruritus of cholestasis. Hepatology. 1999;29:1003-6. 【4.3-12】
7) Bergasa NV. Pruritus and fatigue in primary biliary cirrhosis. Clin Liver Dis. 2003;7:879-900. 【4.3-13】
8) Gillespie DA, Vickers CR. Pruritus and cholestasis: therapeutic options. J Gastroenterol Hepatol.
1993;8:168-73. 【4.3-14】
9) Mela M, Mancuso A, Burroughs AK. Review article: pruritus in cholestatic and other liver diseases. Aliment Pharmacol Ther. 2003;17:857-70. 【4.3-15】
10) Thornton JR, Losowsky MS. Plasma leucine enkephalin is increased in liver disease. Gut.
1989;30:1392-5. 【4.3-17】
11) Thornton JR, Losowsky MS. Opioid peptides and primary biliary cirrhosis. BMJ. 1988;297:1501-4. 【4.3-18】
12) 川島由美. 原発性胆汁性肝硬変におけるそう痒の原因物質に関する研究―opioid peptide の関
与―. 帝京医学雑誌. 2005;28:89-97. 【4.3-19】
13) Bergasa NV, Alling DW, Talbot TL, Swain MG, Yurdaydin C, Turner ML, et al. Effects of naloxone infusions in patients with the pruritus of cholestasis. A double-blind, randomized, controlled trial. Ann Intern Med. 1995;123:161-7. 【4.3-20】
14) Wolfhagen FH, Sternieri E, Hop WC, Vitale G, Bertolotti M, Van Buuren HR. Oral naltrexone
treatment for cholestatic pruritus: a double-blind, placebo-controlled study. Gastroenterology. 1997;113:1264-9. 【4.3-4】
15) Terg R, Coronel E, Sordá J, Muñoz AE, Findor J. Efficacy and safety of oral naltrexone treatment for
pruritus of cholestasis, a crossover, double blind, placebo-controlled study. J Hepatol. 2002;37:717-22. 【4.3-5】
16) Pan ZZ. μ-Opposing actions of the κ-opioid receptor. Trends Pharmacol Sci. 1998;19:94-8. 【4.3-22】
- 7 -

ナルフラフィン塩酸塩 2.6.1 緒言 Page 8
17) Togashi Y, Umeuchi H, Okano K, Ando N, Yoshizawa Y, Honda T, et al. Antipruritic activity of the κ-opioid receptor agonist, TRK-820. Eur J Pharmacol. 2002;435:259-64. 【4.3-54】
- 8 -

ナルフラフィン塩酸塩
医薬品製造販売承認申請書添付資料
2.6.2 薬理試験の概要文
東レ・メディカル株式会社
- 9 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 2
目次
2.6.2 薬理試験の概要文 ................................................................................................................. 8 2.6.2.1 まとめ ............................................................................................................................ 8 2.6.2.2 効力を裏付ける試験 ................................................................................................... 13 2.6.2.3 副次的薬理試験 ........................................................................................................... 54 2.6.2.4 安全性薬理試験 ........................................................................................................... 55 2.6.2.5 薬力学的薬物相互作用試験 ....................................................................................... 61 2.6.2.6 考察及び結論 ............................................................................................................... 64 2.6.2.7 図表 .............................................................................................................................. 74 2.6.2.8 参考文献....................................................................................................................... 75
- 10 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 3
表一覧
表 2.6.2-1. TRK-820 のマウスオピオイド受容体選択性 ................................................................ 14 表 2.6.2-2. TRK-820 のモルモットオピオイド受容体選択性 ........................................................ 15 表 2.6.2-3. ヒトオピオイド受容体結合性 ........................................................................................ 15 表 2.6.2-4. TRK-820 および既存オピオイド受容体作動薬のヒトオピオイド受容体作動性 .... 19 表 2.6.2-5. 炎症性メディエーター遊離もしくは分泌および NOS 活性に対する作用 .............. 32 表 2.6.2-6. 結合試験において用いた特異的標識リガンドおよび標本 ........................................ 34 表 2.6.2-7. 各種受容体、イオンチャネルおよびトランスポーターへの特異的標識リガンド
結合に対する TRK-820 の阻害作用 ............................................................................... 37 表 2.6.2-8. 経口剤中の不純物のオピオイド受容体結合性(Ki 値) ........................................... 43 表 2.6.2-9. 代謝物のオピオイド受容体結合性(プライマリアッセイ) .................................... 44 表 2.6.2-10. 代謝物のオピオイド受容体結合性(Ki 値) ............................................................... 44 表 2.6.2-11. 経口剤中の不純物および代謝物のヒトオピオイド受容体作動性 ............................ 47 表 2.6.2-12. 結合試験において用いた特異的標識リガンドおよび標本 ........................................ 48 表 2.6.2-13. 各種受容体、イオンチャネルおよびトランスポーターへの特異的標識リガンド
結合に対する TRK-820 の代謝物の阻害作用 ............................................................... 51 表 2.6.2-14. TRK-820 の麻酔下イヌにおける平均血圧への影響 .................................................... 60 表 2.6.2-15. イヌにおける単回経口投与時の TRK-820 の Cmax ....................................................... 72
- 11 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 4
図一覧
図 2.6.2-1. TRK-820 および既存オピオイド受容体作動薬のヒトオピオイド受容体作動性 .... 18 図 2.6.2-2. ヒスタミン誘発引っ掻き行動に対する作用 ................................................................ 21 図 2.6.2-3. サブスタンス P 誘発引っ掻き行動に対する作用 ........................................................ 23 図 2.6.2-4. デオキシコール酸誘発引っ掻き行動に対する作用 .................................................... 25 図 2.6.2-5. モルヒネ大槽内投与誘発引っ掻き行動に対する作用 ................................................ 27 図 2.6.2-6. 皮膚炎自然発症 NC/Nga 系マウスの引っ掻き行動に対する作用 ............................. 28 図 2.6.2-7. 引っ掻き行動抑制作用の持続時間 ................................................................................ 29 図 2.6.2-8. 引っ掻き行動抑制作用における耐性の形成能 ............................................................ 31 図 2.6.2-9. 引っ掻き行動抑制作用に対するオピオイド κ 受容体拮抗薬の皮下投与の影響 .... 40 図 2.6.2-10. 引っ掻き行動抑制作用に対するオピオイド κ受容体拮抗薬の脳室内投与の影響
............................................................................................................................................ 41 図 2.6.2-11. 局所麻酔作用の評価 ........................................................................................................ 42 図 2.6.2-12. 経口剤中の不純物および代謝物のヒトオピオイド受容体作動性 ............................ 46 図 2.6.2-13. 代謝物のサブスタンス P 誘発引っ掻き行動に対する作用 ........................................ 54 図 2.6.2-14. 中枢神経抑制作用におけるケトチフェンとの相互作用 ............................................ 62 図 2.6.2-15. 中枢神経抑制作用におけるニトラゼパムとの相互作用 ............................................ 63
- 12 -
ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 5
用語および略語一覧
用語および略語 説明
10α-OH TRK-820 遊離塩基の 10 位 α 水酸基化体(TRK-820 経口剤中の不純
物) 5-HT 5-Hydroxytriptamine(セロトニン)
8-OH-DPAT 8-Hydroxy-2(di-n-propylamino)-tetraline(セロトニン 5-HT1A 受容体作
動薬) A23187 カルシウムイオノフォア AMPA α-Amino-3-hydroxy-5-methyl-4-isoxazole propionic acid ANP 心房性利尿ペプチド(Atrial natriuretic peptide) APD20 Action potential duration at 20% repolarization APD50 Action potential duration at 50% repolarization APD90 Action potential duration at 90% repolarization APT Aminopotentidine(ヒスタミン H2受容体拮抗薬) ATP アデノシン 3 リン酸(Adenosine triphosphate) AVP アルギニンバソプレッシン(Arginin vasopressin) cAMP 環状アデノシン 1 リン酸(Cyclic adenosine monophosphate) CGRP カルシトニン遺伝子関連ペプチド(Calcitonin gene-related peptide) Cmax 最高血漿(血清)中濃度(Maximum plasma (serum) concentration)
CMC カルボキシメチルセルロースナトリウム塩(Carboxymethylcellulose sodium salt)
DAMGO [D-Ala2, N-Me-Phe4, Gly5-ol]-Enkephalin(オピオイド μ 受容体完全作
動薬)の酢酸塩 de-CPM TRK-820 遊離塩基の脱シクロプロピルメチル体(TRK-820 の代謝物) de-CPM·T TRK-820 遊離塩基の脱シクロプロピルメチル体の酒石酸塩
de-CPM-G TRK-820 遊離塩基の脱シクロプロピルメチル体のグルクロン酸抱合
体(TRK-820 の代謝物) DMSO ジメチルスルホキシド(Dimethylsulfoxide)
Dose ratio マウス摘出輸精管およびモルモット摘出回腸を用いたオピオイド受
容体選択性試験(in vitro)において、TRK-820 の濃度-反応回帰直線
が何倍平行移動したかを示した値(平行線検定) DPCPX 8-Cyclopentyl-1,3-dipropylxanthine(アデノシン A1 受容体拮抗薬) DPDPE [D-Pen2,5]-Enkephalin(オピオイド δ 受容体完全作動薬) DTG 1,3-Di-(2-tolyl)-guanidine(シグマ受容体リガンド) EC50 50%有効濃度(50% Effective concentration) ED50 50%有効投与量(50% Effective dose) eq. 等量(Equivalence) GABA γ-アミノ酪酸(γ-Aminobutyric acid) GPI モルモット回腸(Guinea pig ileum) hERG ヒト ether-a-go-go 関連遺伝子(Human ether-a-go-go related gene) i.c. 大槽内:小脳延髄槽(Intracisternal) IC50 50%阻害濃度(50% Inhibitory concentration) i.c.v. 脳室内(Intracerebroventricular) i.d. 皮内(Intradermal) IL-1β インターロイキン-1β(Interleukin-1β) IL-2 インターロイキン-2(Interleukin-2)
- 13 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 6
用語および略語一覧(続き)
用語および略語 説明 IL-6 インターロイキン-6(Interleukin-6) IL-8 インターロイキン-8(Interleukin-8)
Imax Maximal inhibitory rate(フォルスコリン刺激誘発 cAMP 産生に対す
る抑制作用試験における最大抑制率) i.p. 腹腔内(Intraperitoneal) i.v. 静脈内(Intravenous) L-NMMA NG-Monomethyl-L-arginine(一酸化窒素合成酵素阻害薬) LPS Lipopolysaccharide(エンドトキシン) LTD4 ロイコトリエン D4(Leukotriene D4) MCP-1 Monocyte chemoattractant protein-1 MIP-1α Macrophage inflammatory protein-1α MVD マウス輸精管(Mouse vas deferens) NFA-G TRK-820 遊離塩基のグルクロン酸抱合体(TRK-820 の代謝物) NKA Neurokinin A NKB Neurokinin B NMDA N-Methyl-D-aspartic acid NO 一酸化窒素(Nitric oxide)
nor-BNI ノルビナルトルフィミン 2 塩酸塩 1 水和物(オピオイド κ 受容体拮
抗薬、Nor-binaltorphimine dihydrochloride monohydrate) NOS 一酸化窒素合成酵素(Nitric oxide synthase)
NTI ナルトリンドールメタンスルホン酸塩(オピオイド δ 受容体拮抗薬、
Naltrindole methanesulfonate) ORL1 Opioid receptor-like 1 PAF 血小板活性化因子(Platelet activating factor) PBC 原発性胆汁性肝硬変(Primary Biliary Cirrhosis) PBS リン酸緩衝生理食塩水(Phosphate buffered saline) pCO2 炭酸ガス分圧 PGD2 プロスタグランジン D2(Prostaglandin D2) PGE2 プロスタグランジン E2(Prostaglandin E2) PGI2 プロスタサイクリン(Prostacyclin) pH 水素イオン濃度 p.o. 経口(Per os, Per oral) pO2 酸素分圧 QTc 補正 QT 間隔(Corrected QT interval) s.c. 皮下(Subcutaneous) SPF 特定病原体不在の(Specific pathogen free) TBPS t-Butylbicyclophosphorothionate(GABAA受容体 Cl-チャネル阻害薬) TCP Tenocyclidine(NMDA 受容体拮抗薬) TNF-α 腫瘍壊死因子(Tumor necrosis factor-α)
U-69593 (+)-(5α, 7α, 8β)-N-Methyl-N-[7-(1-pyrrolidinyl)-1-oxaspiro[4, 5]dec-8- yl]-benzeneacetamide(オピオイド κ 受容体完全作動薬)
VIP 血管作動性腸管ペプチド(Vasoactive intestinal peptide) β-エンドルフィン オピオイド μ 受容体作動性を有する内因性オピオイドペプチド A7r5 細胞 ラット血管平滑筋由来細胞(Rat aortic smooth muscle cell line)
- 14 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 7
用語および略語一覧(続き)
用語および略語 説明 AtT-20 細胞 下垂体由来腫瘍細胞 Balb/c 3T3 細胞 マウス線維芽由来細胞(Mouse fibroblast cell line) Child-Pugh 分類 肝疾患の重症度分類
CHO 細胞 チャイニーズハムスター卵巣由来細胞(Chinese hamster ovary cell line)
HEK-293 細胞 ヒト胎児腎臓由来細胞(Human embryonic kindney cell line) HL-60 細胞 ヒト前骨髄性白血病細胞(Human promyelocytic leukemia cell line) HT-29 細胞 ヒト結腸腺癌細胞(Human colon adenocarcinoma cell line)
HUVEC 細胞 ヒト臍帯静脈由来血管内皮細胞(Human umbilical vein endothelial cell line)
Ke 値
マウス摘出輸精管およびモルモット摘出回腸を用いたオピオイド受
容体選択性試験(in vitro)において、TRK-820 の濃度-反応回帰直線
を高用量側に 2 倍平行移動させるのに必要な拮抗薬の濃度 Ke (nmol/L)=[拮抗薬の濃度 (nmol/L)]/[Dose ratio-1]
Ki 値 受容体結合試験における結合阻害定数 Leu-エンケファリン オピオイド μ 受容体作動性を有する内因性オピオイドペプチド LLC-PK1 細胞 ブタ腎上皮由来細胞(Porcine kidney epithelial cell line) Met-エンケファリン オピオイド μ 受容体作動性を有する内因性オピオイドペプチド RAW264-7 細胞 マウスマクロファージ様細胞(Mouse monocyte-macrophage cell line) SK-N-MC 細胞 ヒト神経芽細胞腫由来細胞(Human neuroblastoma cell line) THP-1 細胞 ヒト単球由来細胞(Human monocyte cell line) U937 細胞 ヒト単球性白血病細胞(Human monoblastic leukemia cell line) エチニルエストラジオール 17α-エチニルエストラジオール(卵胞ホルモン薬) エンドモルフィン-1 オピオイド μ 受容体作動性を有する内因性オピオイドペプチド オキサトミド オキサトミド(抗ヒスタミン薬) クロルフェニラミン クロルフェニラミンマレイン酸塩(抗ヒスタミン薬) ケトチフェン ケトチフェンフマル酸塩(抗ヒスタミン薬) コンパウンド 48/80 ヒスタミン遊離物質 サブスタンス P サブスタンス P 酢酸塩水和物(起痒剤) デオキシコール酸 デオキシコール酸ナトリウム 1 水和物(起痒剤) ナルトレキソン ナルトレキソン塩酸塩(オピオイド μ 受容体拮抗薬) ナロキソン ナロキソン塩酸塩(オピオイド μ 受容体拮抗薬) ニトラゼパム ニトラゼパム(睡眠導入薬) ヒスタミン ヒスタミン(起痒剤)
ブトルファノール ブトルファノール酒石酸塩(オピオイド κ 受容体作動性を有する麻
薬拮抗性鎮痛薬)
ブプレノルフィン ブプレノルフィン塩酸塩(オピオイド μ 受容体部分作動性を有する
麻薬拮抗性鎮痛薬) プロカイン プロカイン塩酸塩(局所麻酔薬) ヘモグロビン O2 saturation ヘモグロビン酸素飽和濃度 ペントバルビタール ペントバルビタールナトリウム モルヒネ モルヒネ塩酸塩(麻薬性鎮痛薬)
- 15 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 8
2.6.2 薬理試験の概要文
2.6.2.1 まとめ
TRK-820 のオピオイド受容体に対する in vitro の作用、マウスの各種そう痒モデルの引っ掻き行
動を指標に in vivo での止痒作用を評価した。さらに、TRK-820 の止痒作用の発現機序を in vitro
および in vivo の各試験により評価した。慢性肝疾患に伴うそう痒症には複数の因子が関与してい
ると考えられており、抗ヒスタミン薬などの既存治療薬に抵抗性を示す患者の存在が知られてい
る 1, 2)。また、痒みを有する肝疾患患者の血漿中で内因性オピオイドペプチドの増加が認められる
こと 3-5)や、オピオイド μ 受容体拮抗薬が肝疾患に伴うそう痒に対して有効であること 6-8)などか
ら、肝疾患患者の痒みの発現には、オピオイド μ 受容体の活性化が関与していることが示唆され
る。したがって、TRK-820 の止痒作用は、起痒剤であるヒスタミンおよびサブスタンス P の皮内
投与によるマウスの引っ掻き行動、胆汁うっ滞性のそう痒モデルであるデオキシコール酸の皮内
投与によるマウスの引っ掻き行動、オピオイド μ 受容体作動薬であるモルヒネの大槽内投与によ
るマウスの引っ掻き行動および自然発症性のアトピー性皮膚炎マウスの引っ掻き行動に対する抑
制作用を指標に、抗ヒスタミン薬感受性および抵抗性のそう痒モデルに対する作用を網羅的に検
討することにより評価した。なお、TRK-820 の経口投与によるバイオアベイラビリティが、マウ
ス(32%)と比較してラット(4.6%)で非常に低く、さらに、痒みの動物モデルのほとんどがマ
ウスを用いて構築され、一般によく使用されていることから主薬効試験の動物種はマウスを選択
した。
上記試験に加えて、TRK-820 の安全性薬理試験については、「新医薬品等の製造(輸入)承認申
請に必要な一般薬理試験のガイドラインについて」(平成 3 年 1 月 29 日 薬新薬第 4 号)もしく
は「安全性薬理試験ガイドラインについて」(平成 13 年 6 月 21 日 医薬審発第 902 号)に準じて、
安全性薬理コアバッテリー試験、安全性薬理コアバッテリーに対するフォローアップ試験および
補足的安全性薬理試験を実施した。
また、中枢抑制作用に対する薬力学的薬物相互作用に関して、抗ヒスタミン薬もしくは睡眠導
入薬と TRK-820 の併用について評価した。
2.6.2.1.1 効力を裏付ける試験 (1) in vitro
TRK-820 のげっ歯類オピオイド受容体選択性は、マウス摘出輸精管標本およびモルモット摘出
回腸標本の電気刺激収縮を指標にした受容体選択性試験により評価した。いずれの評価において
も TRK-820 はオピオイド κ 受容体に対する選択性が高かった。
また、TRK-820 のヒトオピオイド受容体に対する選択性は、受容体結合性試験およびフォルス
コリン刺激誘発 cAMP 産生に対する抑制作用を指標にした作動性試験により検討した。
- 16 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 9
ヒトオピオイド κ、μ および δ 受容体結合性試験における TRK-820 の結合阻害定数(Ki 値)は、
それぞれ 0.244、2.21 および 484 nmol/L であり、オピオイド κ 受容体に対する結合性は、オピオ
イド μ および δ 受容体と比較して、それぞれ 9 および 1980 倍強いことが示された。
ヒトオピオイド受容体作動性試験では、ヒトオピオイド κ 受容体発現細胞においてフォルスコ
リン刺激誘発 cAMP 産生に対する TRK-820 の最大抑制率(Imax)は 91%であり、標準的な完全作
動薬である U-69593(Imax:91%)と同等であった。ヒトオピオイド μ 受容体発現細胞では TRK-820
の Imaxは 53%であり、標準的な完全作動薬である DAMGO(Imax:77%)およびモルヒネ(Imax:
76%)と比較して統計学的に有意に低値であったが、部分作動薬であるブプレノルフィン(Imax:
56%)およびブトルファノール(Imax:47%)と同程度であった。ヒトオピオイド δ 受容体発現細
胞では TRK-820 の Imaxは 78%であり、標準的な完全作動薬である DPDPE(Imax:87%)およびブ
トルファノール(Imax:83%)と比較して統計学的に有意に小さく、ブプレノルフィン(Imax:80%)
と同程度であった。さらに、オピオイド κ、μ、δ 受容体に対する作動性について、EC50の比は、
モルヒネで 1:0.1:1.0、ブプレノルフィンで 1:0.4:0.6、ブトルファノールで 1:4.4:6.5、TRK-820
で 1:203:2610 であった。したがって、TRK-820 は、高活性のオピオイド κ 受容体完全作動薬で
あり、オピオイド μ 受容体に対して部分作動性を有し、オピオイド δ 受容体作動性は弱いと考え
られた。
以上のことから、TRK-820 は、オピオイド κ 受容体に対する結合性が強く、さらに、既存のオ
ピオイド受容体作動薬(モルヒネ、ブプレノルフィンおよびブトルファノール)との比較から、
既存薬と明確に異なるプロファイルを有し、オピオイド κ 受容体に対する作動性が非常に強い、
オピオイド κ 受容体選択的完全作動薬であることが示唆された。
(2) in vivo
各種起痒剤をマウスの吻側背部皮内に投与することによって誘発される引っ掻き行動に対する
TRK-820 の作用を評価した。
抗ヒスタミン薬が有効なヒスタミン皮内投与誘発引っ掻き行動は、TRK-820 の経口投与によっ
て用量依存的に抑制され、ED50は 7.30 μg/kg であった。したがって、TRK-820 は抗ヒスタミン薬
が有効な痒みに対して止痒作用を有する可能性が示唆された。
抗ヒスタミン薬が十分な効果を示さないサブスタンス P 皮内投与誘発引っ掻き行動、抗ヒスタ
ミン薬が無効なデオキシコール酸誘発引っ掻き行動(胆汁うっ滞性の痒みモデル)、モルヒネ大槽
内投与誘発引っ掻き行動(中枢神経系のオピオイド μ 受容体の活性化に伴う痒みモデル)および
NC/Nga 系マウスをコンベンショナル環境下で飼育し、アトピー性皮膚炎を発症させたマウスに認
められる引っ掻き行動は、TRK-820 によって用量依存的に抑制され、ED50はそれぞれ、19.6 μg/kg
(経口投与)、7.62 μg/kg(経口投与)、2.34 μg/kg(皮下投与)および 46.1 μg/kg(経口投与)であ
- 17 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 10
った。したがって、TRK-820 は、抗ヒスタミン薬などの既存治療薬に抵抗性の痒みに対して止痒
作用を有する可能性が示唆された。
サブスタンス P 皮内投与誘発引っ掻き行動において、TRK-820(100 μg/kg)経口投与による止
痒作用の持続時間は約 6 時間であった。
TRK-820(100 μg/kg)の 1 日 2 回、7 日間反復経口投与によって、サブスタンス P 皮内投与誘
発引っ掻き行動抑制作用の ED50は、1.8 倍増加したのみであったことから、止痒作用において強
い耐性を形成する可能性は低いと考えられた。
(3) 作用機序
以下の in vitro および in vivo の各試験の結果から、TRK-820 の止痒作用は、未変化体が中枢神経
系のオピオイド κ 受容体を活性化することによって発現している可能性が示唆されている。
TRK-820 は、in vitro において炎症性メディエーター遊離もしくは分泌(ヒスタミン遊離、TNF-α
分泌、IL-1β 分泌、IL-6 分泌、PGE2 分泌および PGD2 分泌)に対する抑制作用を示さず、また、
誘導型 NOS および構成型 NOS の活性に対する阻害作用も示さなかった。
イ TRK-820 のオピオド受容体以外の種々の受容体、イオンチャネルおよびトランスポーターに
対する結合性を in vitro にて評価したところ、ムスカリン M1受容体に対して、Ki 値は 1700 nmol/L
であった。また、TRK-820 は 1000 nmol/L で、オルファニン ORL1 受容体への特異的標識リガンド
である[3H]ノシセプチンの結合を 47%阻害した。しかし、TRK-820 のオピオイド κ 受容体に対す
る Ki 値が 0.244 nmol/L であることから、いずれもオピオイド κ 受容体に対する結合性と比較して
著しく低いと考えられた。なお、TRK-820 はその他の受容体、イオンチャネルおよびトランスポ
ーターに対して、ほとんど結合性を示さなかった。
TRK-820 経口投与によるサブスタンス P 皮内投与誘発マウス引っ掻き行動の抑制作用は、オピ
オイド κ 受容体拮抗薬 nor-BNI の皮下投与により部分的に拮抗され、脳室内投与により完全に消
失した。さらに、TRK-820 はモルモットにおいて局所麻酔作用を示さなかった。
TRK-820 経口剤(軟カプセル剤)中の不純物である 10α-OH、代謝物である de-CPM、NFA-G お
よび de-CPM-G は、in vitro のヒトオピオイド κ、μ および δ 受容体結合試験および作動性試験の結
果から、いずれのオピオイド受容体に対しても TRK-820 より結合性および作動性が低いことが示
唆された。また、de-CPM、NFA-G および de-CPM-G はその他の受容体、イオンチャネルおよびト
ランスポーターに対して、ほとんど結合性を示さなかった。これに加えて、サブスタンス P 皮内
投与誘発マウス引っ掻き行動に対して、de-CPM·T(de-CPM の酒石酸塩)、NFA-G および de-CPM-G
を、TRK-820 が皮下投与により統計学的に有意な抑制作用を示す用量(10 μg/kg)の 100 倍量
(1000 μg/kg)まで皮下投与しても抑制作用を発現しなかった。
- 18 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 11
2.6.2.1.2 安全性薬理試験
(1) 安全性薬理コアバッテリー試験
Irwin 法を用いたラットの一般症状観察において、経口投与した TRK-820 は、身づくろいの低
下、自発運動の低下、体温の低下、眼瞼下垂、警戒性の低下、反応性の低下、よろめき歩行、四
肢伸長、流涙、逃避反応の低下、痛覚反応の低下、耳介反射の低下、腹這い姿勢、異常歩行およ
び縮瞳という主に中枢神経系の抑制作用に基づくと考えられる一般症状の変化を引き起こしたが、
ほとんど軽度であった。
hERG 遺伝子を導入した HEK-293 細胞において hERG 電流に対する TRK-820 の IC50 は、
Child-Pugh 分類グレード A の代償性肝硬変患者および Child-Pugh 分類グレード B の肝硬変患者に
おける予想臨床使用最高用量 5 μg/body の反復経口投与時の推定 Cmaxと比較して、それぞれ約
63000 および 29000 倍高濃度であった。
無麻酔・非拘束イヌにおいて、TRK-820 を経口投与したところ、血圧低下および心拍数の増加
が認められたが、心電図もしくは呼吸系への影響は認められなかった。TRK-820 のイヌにおける
QTc もしくは呼吸系に対する無作用量の Cmaxは、Child-Pugh 分類グレード A の代償性肝硬変患者
および Child-Pugh 分類グレード B の肝硬変患者における予想臨床使用最高用量(5 μg/body)の反
復経口投与時の推定 Cmax のそれぞれ約 240 および 110 倍以上高濃度であった。さらに、血圧およ
び心拍数を含めた心血管系に対する無作用量の Cmaxは、Child-Pugh 分類グレード A の代償性肝硬
変患者および Child-Pugh 分類グレード B の肝硬変患者における予想臨床使用最高用量(5 μg/body)
の反復経口投与時の推定 Cmaxのそれぞれ約 8 および 4 倍高濃度であった。
(2) 安全性薬理コアバッテリーに対するフォローアップ試験
アカゲザルを用いた一般症状観察において、静脈内投与した TRK-820 は、運動低下、うずくま
り姿勢、観察者への攻撃行動の増強、観察者への攻撃行動の減弱、腹臥位、動作緩慢、観察者へ
の怯え表情の減弱、閉眼、流涎、口の半開状態および運動失調を引き起こした。なお、この一般
症状の変化は止痒作用が認められる用量よりも高用量で発現すると考えられた。
TRK-820 は、マウスおよびラットの移所運動を経口投与により減少させた。また、マウスの回
転かご試験でも、TRK-820 は経口投与および皮下投与により自発運動抑制作用を発現した。また、
マウスのロタロッド試験において、TRK-820 は経口投与により、協調運動を阻害した。さらに、
経口投与した TRK-820 は、マウスのペントバルビタール誘発睡眠の持続時間(睡眠時間)に対し
て延長作用を示した。これらのマウスへの作用は、止痒作用が認められる用量よりも 5~49 倍高
用量で発現すると考えられた。
皮下投与した TRK-820 は、止痒作用が発現すると考えられる用量とほぼ同じ用量から、ラット
の新皮質前頭葉の脳波の周波数解析に対して、アルファ帯域およびベータ-1 帯域の減少、海馬の
- 19 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 12
周波数解析に対しては、アルファ帯域の減少およびベータ-2 帯域の増加を引き起こした。睡眠-
覚醒周期の各脳波水準に対しては、覚醒期の増加、徐波睡眠期の減少、速波睡眠期の減少、徐波
睡眠期の潜時時間の延長および速波睡眠期の潜時時間の延長を引き起こした。
TRK-820 は、マウス酢酸ライジング試験において経口および皮下投与により、止痒作用が発現
すると考えられる用量と同程度もしくは高い用量で鎮痛作用を示すことが示唆された。
TRK-820 は経口投与により、マウスにおいて痙攣誘発および抗痙攣作用を示さなかった。
経口投与した TRK-820 は、ラットの体温に対して、比較的高用量で体温低下を引き起こした。
TRK-820 のモルモット摘出乳頭筋の活動電位に対する無影響濃度は、Child-Pugh 分類グレード
A の代償性肝硬変患者および Child-Pugh 分類グレード B の肝硬変患者における予想臨床使用最高
用量(5 μg/body)の反復経口投与時の推定 Cmaxのそれぞれ約 23000 および 10000 倍高濃度であっ
た。イソフルラン麻酔イヌを用いた心血管系の試験において、TRK-820 の静脈内投与により、血
圧低下が認められ、心拍数が減少傾向を示した。一方、心電図への影響は認められなかった。
TRK-820 の麻酔イヌにおける QTc に対する無作用量の投与終了直後の血漿中 TRK-820 濃度は、
Child-Pugh 分類グレード A の代償性肝硬変患者および Child-Pugh 分類グレード B の肝硬変患者に
おける予想臨床使用最高用量(5 μg/body)の反復経口投与時の推定 Cmaxのそれぞれ約 890 および
410 倍高濃度であった。
(3) 補足的安全性薬理試験
経口投与した TRK-820 は、ラットにおいて尿量の増加、尿中 Na+総排泄量の減少、尿中 K+総排
泄量の増加および尿中 Cl-総排泄量の減少を引き起こした。
モルモット摘出回腸のアセチルコリン、ヒスタミンおよび塩化バリウム刺激による収縮に対し
て、TRK-820 は影響を及ぼさなかった。
TRK-820 は、マウス腸管輸送能に対して、止痒作用が発現すると考えられる用量よりも 177~
475 倍高用量で抑制作用を示した。また、その作用は対照薬のモルヒネと比較して弱かった。
2.6.2.1.3 薬力学的薬物相互作用試験
マウスのペントバルビタール誘発睡眠に対する延長作用を指標に抗ヒスタミン薬ケトチフェン
および睡眠導入薬ニトラゼパムとの中枢抑制作用における相互作用を検討した結果、TRK-820 は
ケトチフェンと相加的に作用し、また、ニトラゼパムの作用を用量依存的に増強する可能性が示
唆された。
- 20 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 13
2.6.2.2 効力を裏付ける試験
2.6.2.2.1 受容体選択性(in vitro)
(1) げっ歯類オピオイド受容体選択性
TRK-820 のげっ歯類オピオイド受容体に対する選択性を、マウス摘出輸精管標本およびモルモ
ット摘出回腸標本の電気刺激収縮を指標にした受容体選択性試験により評価した。
1) マウス摘出輸精管(MVD)(2.6.3.2 (1)、4.2.1.1-1: 試験)
a) 方法
ICR 系雄性マウスの輸精管を摘出し、Krebs 栄養液(Mg2+非添加)を満たしたマグヌス装置にセ
ットした。電気刺激(頻度 0.1 Hz、持続 1 msec)により誘発される等尺張力の変化を isometric
transducer を介して測定した。オピオイド κ 受容体拮抗薬 nor-BNI(1、3、10 および 30 nmol/L)、
オピオイド μ 受容体拮抗薬ナロキソン(10、30 および 100 nmol/L)もしくはオピオイド δ 受容体
拮抗薬 NTI(3、10 および 30 nmol/L)は TRK-820 を添加する 15 分前に加えた。TRK-820 は 0.02
~0.54 nmol/L(拮抗薬非存在下)、0.02~4.86 nmol/L(nor-BNI 1 nmol/L 存在下)、0.02~14.58 nmol/L
(nor-BNI 3 nmol/L 存在下)、0.02~131.22 nmol/L(nor-BNI 10 および 30 nmol/L 存在下)、0.02~
1.62 nmol/L(ナロキソン 10、30 および 100 nmol/L 存在下)、0.02~0.54 nmol/L(NTI 3 および
10 nmol/L 存在下)および 0.02~1.62 nmol/L(NTI 30 nmol/L 存在下)の濃度範囲を用いた。なお、
実験は 2 回に分けて実施し、第 1 回目に nor-BNI を用いたオピオイド κ 受容体に対する選択性、
第 2 回目にナロキソンもしくは NTI を用いたオピオイド μ もしくは δ 受容体に対する選択性を評
価した。
b) 成績
TRK-820 は、MVD 標本の電気刺激誘発収縮運動を濃度依存的に抑制し、その IC50 は第 1 回目
の実験では 0.080 nmol/L(95%信頼区間:0.067~0.095 nmol/L)および第 2 回目の実験では
0.12 nmol/L(95%信頼区間:0.063~0.24 nmol/L)であった。
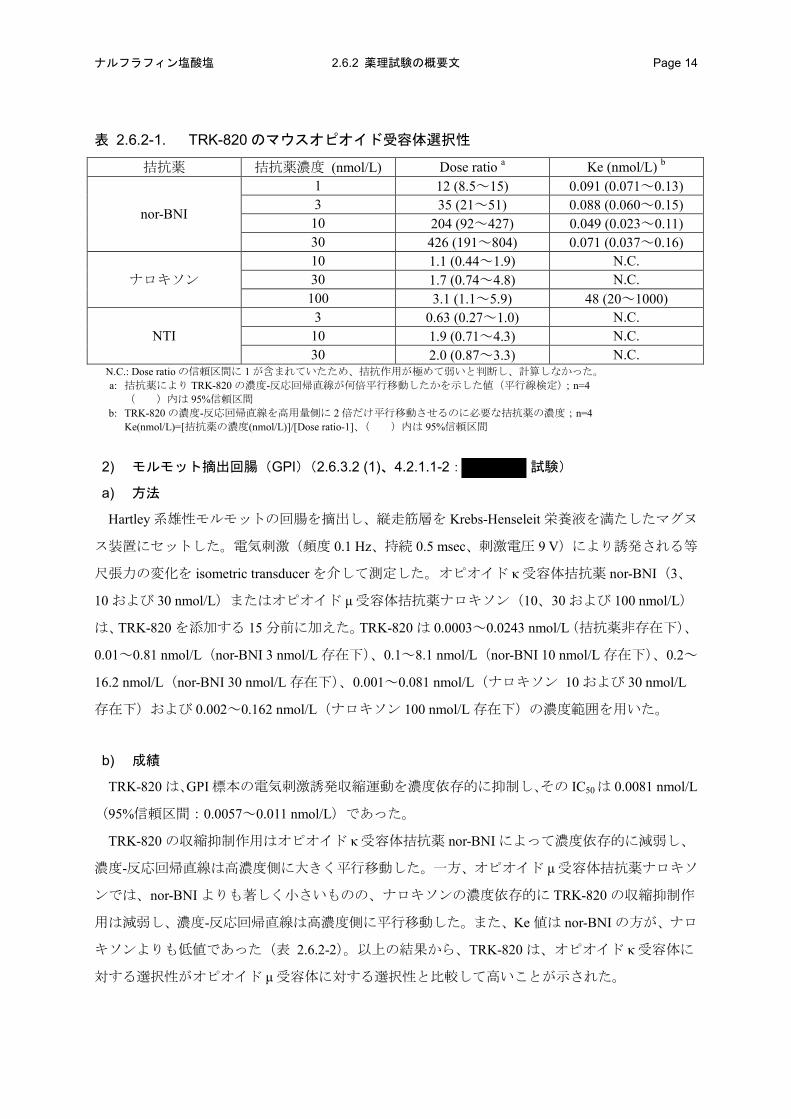
TRK-820 の収縮抑制作用はオピオイド κ 受容体拮抗薬 nor-BNI によって濃度依存的に減弱し、
濃度-反応回帰直線は高濃度側に大きく平行移動した。一方、オピオイド μ 受容体拮抗薬ナロキソ
ンおよびオピオイド δ 受容体拮抗薬 NTI では、TRK-820 の収縮抑制作用は減弱せず、濃度-反応回
帰直線はほとんど変化しなかった(表 2.6.2-1)。以上の結果から、TRK-820 はオピオイド κ 受容
体に対する選択性がオピオイド μおよび δ受容体に対する選択性と比較して高いことが示された。
- 21 -
ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 14
表 2.6.2-1. TRK-820 のマウスオピオイド受容体選択性
拮抗薬 拮抗薬濃度 (nmol/L) Dose ratio a Ke (nmol/L) b
nor-BNI
1 12 (8.5~15) 0.091 (0.071~0.13) 3 35 (21~51) 0.088 (0.060~0.15) 10 204 (92~427) 0.049 (0.023~0.11) 30 426 (191~804) 0.071 (0.037~0.16)
ナロキソン 10 1.1 (0.44~1.9) N.C. 30 1.7 (0.74~4.8) N.C. 100 3.1 (1.1~5.9) 48 (20~1000)
NTI 3 0.63 (0.27~1.0) N.C. 10 1.9 (0.71~4.3) N.C. 30 2.0 (0.87~3.3) N.C.
N.C.: Dose ratio の信頼区間に 1 が含まれていたため、拮抗作用が極めて弱いと判断し、計算しなかった。 a: 拮抗薬により TRK-820 の濃度-反応回帰直線が何倍平行移動したかを示した値(平行線検定);n=4
( )内は 95%信頼区間 b: TRK-820 の濃度-反応回帰直線を高用量側に 2 倍だけ平行移動させるのに必要な拮抗薬の濃度;n=4
Ke(nmol/L)=[拮抗薬の濃度(nmol/L)]/[Dose ratio-1]、( )内は 95%信頼区間
2) モルモット摘出回腸(GPI)(2.6.3.2 (1)、4.2.1.1-2: 試験)
a) 方法
Hartley 系雄性モルモットの回腸を摘出し、縦走筋層を Krebs-Henseleit 栄養液を満たしたマグヌ
ス装置にセットした。電気刺激(頻度 0.1 Hz、持続 0.5 msec、刺激電圧 9 V)により誘発される等
尺張力の変化を isometric transducer を介して測定した。オピオイド κ 受容体拮抗薬 nor-BNI(3、
10 および 30 nmol/L)またはオピオイド μ 受容体拮抗薬ナロキソン(10、30 および 100 nmol/L)
は、TRK-820 を添加する 15 分前に加えた。TRK-820 は 0.0003~0.0243 nmol/L(拮抗薬非存在下)、
0.01~0.81 nmol/L(nor-BNI 3 nmol/L 存在下)、0.1~8.1 nmol/L(nor-BNI 10 nmol/L 存在下)、0.2~
16.2 nmol/L(nor-BNI 30 nmol/L 存在下)、0.001~0.081 nmol/L(ナロキソン 10 および 30 nmol/L
存在下)および 0.002~0.162 nmol/L(ナロキソン 100 nmol/L 存在下)の濃度範囲を用いた。
b) 成績
TRK-820 は、GPI 標本の電気刺激誘発収縮運動を濃度依存的に抑制し、その IC50は 0.0081 nmol/L
(95%信頼区間:0.0057~0.011 nmol/L)であった。
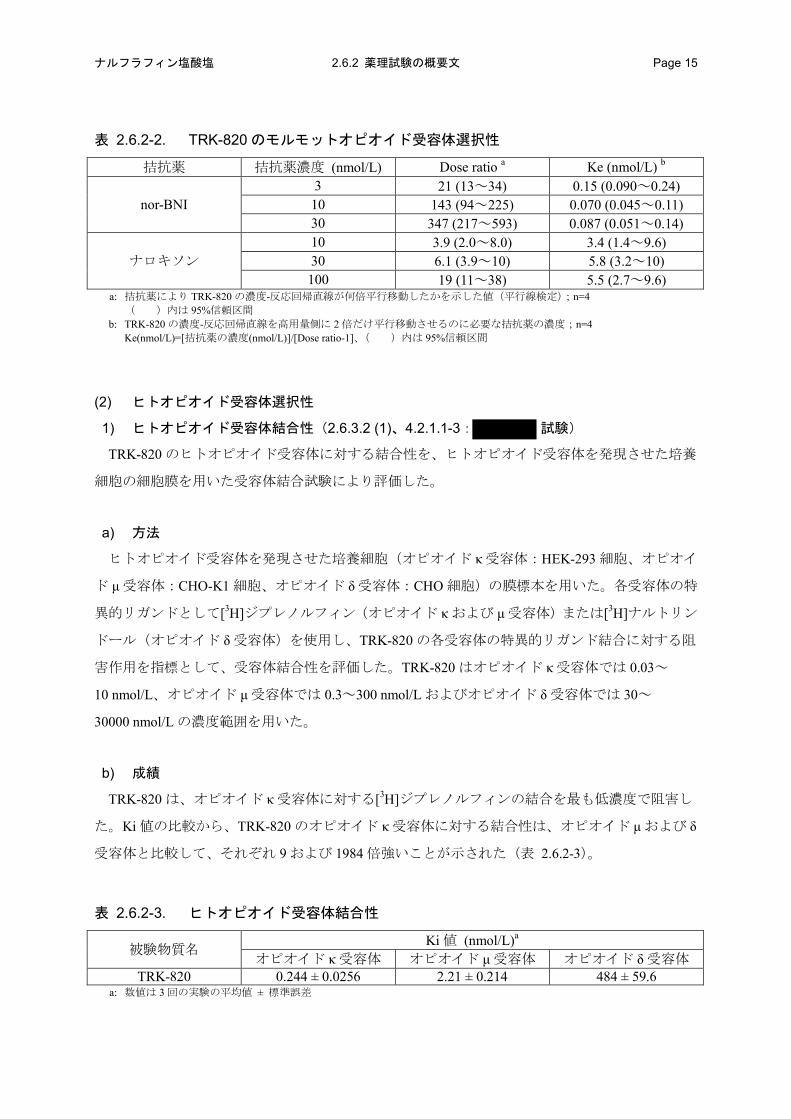
TRK-820 の収縮抑制作用はオピオイド κ 受容体拮抗薬 nor-BNI によって濃度依存的に減弱し、
濃度-反応回帰直線は高濃度側に大きく平行移動した。一方、オピオイド μ 受容体拮抗薬ナロキソ
ンでは、nor-BNI よりも著しく小さいものの、ナロキソンの濃度依存的に TRK-820 の収縮抑制作
用は減弱し、濃度-反応回帰直線は高濃度側に平行移動した。また、Ke 値は nor-BNI の方が、ナロ
キソンよりも低値であった(表 2.6.2-2)。以上の結果から、TRK-820 は、オピオイド κ 受容体に
対する選択性がオピオイド μ 受容体に対する選択性と比較して高いことが示された。
- 22 -
ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 15
表 2.6.2-2. TRK-820 のモルモットオピオイド受容体選択性
拮抗薬 拮抗薬濃度 (nmol/L) Dose ratio a Ke (nmol/L) b
nor-BNI 3 21 (13~34) 0.15 (0.090~0.24) 10 143 (94~225) 0.070 (0.045~0.11) 30 347 (217~593) 0.087 (0.051~0.14)
ナロキソン 10 3.9 (2.0~8.0) 3.4 (1.4~9.6) 30 6.1 (3.9~10) 5.8 (3.2~10) 100 19 (11~38) 5.5 (2.7~9.6)
a: 拮抗薬により TRK-820 の濃度-反応回帰直線が何倍平行移動したかを示した値(平行線検定);n=4 ( )内は 95%信頼区間
b: TRK-820 の濃度-反応回帰直線を高用量側に 2 倍だけ平行移動させるのに必要な拮抗薬の濃度;n=4 Ke(nmol/L)=[拮抗薬の濃度(nmol/L)]/[Dose ratio-1]、( )内は 95%信頼区間
(2) ヒトオピオイド受容体選択性
1) ヒトオピオイド受容体結合性(2.6.3.2 (1)、4.2.1.1-3: 試験)
TRK-820 のヒトオピオイド受容体に対する結合性を、ヒトオピオイド受容体を発現させた培養
細胞の細胞膜を用いた受容体結合試験により評価した。
a) 方法
ヒトオピオイド受容体を発現させた培養細胞(オピオイド κ 受容体:HEK-293 細胞、オピオイ
ド μ 受容体:CHO-K1 細胞、オピオイド δ 受容体:CHO 細胞)の膜標本を用いた。各受容体の特
異的リガンドとして[3H]ジプレノルフィン(オピオイド κ および μ 受容体)または[3H]ナルトリン
ドール(オピオイド δ 受容体)を使用し、TRK-820 の各受容体の特異的リガンド結合に対する阻
害作用を指標として、受容体結合性を評価した。TRK-820 はオピオイド κ 受容体では 0.03~
10 nmol/L、オピオイド μ 受容体では 0.3~300 nmol/L およびオピオイド δ 受容体では 30~
30000 nmol/L の濃度範囲を用いた。
b) 成績
TRK-820 は、オピオイド κ 受容体に対する[3H]ジプレノルフィンの結合を最も低濃度で阻害し
た。Ki 値の比較から、TRK-820 のオピオイド κ 受容体に対する結合性は、オピオイド μ および δ
受容体と比較して、それぞれ 9 および 1984 倍強いことが示された(表 2.6.2-3)。
表 2.6.2-3. ヒトオピオイド受容体結合性
被験物質名 Ki 値 (nmol/L)a
オピオイド κ 受容体 オピオイド μ 受容体 オピオイド δ 受容体 TRK-820 0.244 ± 0.0256 2.21 ± 0.214 484 ± 59.6
a: 数値は 3 回の実験の平均値 ± 標準誤差
- 23 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 16
2) ヒトオピオイド受容体作動性(2.6.3.2 (1)、4.2.1.1-4: 試験)
TRK-820 のヒトオピオイド受容体作動性を、ヒトオピオイド受容体を発現させた CHO 細胞を用
いたフォルスコリン刺激による cAMP 産生に対する抑制作用を指標に評価した。
a) 方法
ヒトオピオイド受容体を発現させた培養細胞(オピオイド κ 受容体:CHO-K1 細胞、オピオイ
ド μ および δ 受容体:CHO-dhfr(-)細胞)を用いた。フォルスコリン 30 μmol/L と共に TRK-820 ま
たは既存のオピオイド受容体作動薬を各細胞に適用し、37°C で 30 分間インキュベーションした。
産生された cAMP 量を AlphaScreenTM cAMP Assay kit および万能マイクロプレートアナライザー
(Fusion α システム)を用いて測定した。TRK-820 は 0.00003~10 nmol/L(オピオイド κ 受容体)
および 0.01~3000 nmol/L(オピオイド μ および δ 受容体)、モルヒネは 0.03~10000 nmol/L(すべ
ての受容体)、ブプレノルフィンは 0.01~3000 nmol/L(すべての受容体)、ブトルファノールは 0.003
~1000 nmol/L(オピオイド κ 受容体)および 0.01~3000 nmol/L(オピオイド μ および δ 受容体)、
U-69593 は 0.001~300 nmol/L(オピオイド κ 受容体)、DAMGO は 0.003~1000 nmol/L(オピオイ
ド μ 受容体)、DPDPE は 0.0003~100 nmol/L(オピオイド δ 受容体)の濃度範囲を用いた。
b) 成績
ヒトオピオイド κ 受容体発現細胞においてフォルスコリン刺激誘発 cAMP 産生に対する
TRK-820 の最大抑制率(Imax)は 91%であり、標準的な完全作動薬である U-69593(Imax:91%)
と同等であった。一方、モルヒネおよびブトルファノールの Imaxはそれぞれ 80および 85%であり、
TRK-820 および U-69593 と比較してわずかに低値を示した。さらに、ブプレノルフィンの Imaxは
48%であり、TRK-820 および U-69593 の約 50%まで低下したが、統計学的に有意な差は認められ
なかった。また、EC50 の比較では、TRK-820(0.00816 nmol/L)< U-69593(0.642 nmol/L)< ブト
ルファノール(0.752 nmol/L)< ブプレノルフィン(4.13 nmol/L)< モルヒネ(391 nmol/L)であ
り、TRK-820 が最も低濃度であった(図 2.6.2-1、表 2.6.2-4)。
ヒトオピオイド μ 受容体発現細胞では TRK-820 の Imaxは 53%であり、標準的な完全作動薬であ
る DAMGO(Imax:77%)およびモルヒネ(Imax:76%)と比較して統計学的に有意に低値であった
が、ブプレノルフィン(Imax:56%)およびブトルファノール(Imax:47%)とほぼ同程度であった。
ただし、ブトルファノールとの間には統計学的に有意な差が認められた。EC50 の比較では、ブプ
レノルフィン(1.59 nmol/L)≈ TRK-820(1.66 nmol/L)< ブトルファノール(3.34 nmol/L)< DAMGO
(5.63 nmol/L)< モルヒネ(35.7 nmol/L)であり、TRK-820 はブプレノルフィンと同程度の EC50
であった(図 2.6.2-1、表 2.6.2-4)。しかしながら、ブプレノルフィンのオピオイド κ 受容体に対
- 24 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 17
する EC50は、オピオイド μ 受容体に対する EC50の 2.6 倍であったが、TRK-820 のオピオイド κ
受容体に対する EC50 は、オピオイド μ 受容体に対する EC50の 1/203 であった。
ヒトオピオイド δ 受容体発現細胞では TRK-820 の Imaxは 78%であり、標準的な完全作動薬であ
る DPDPE(Imax:87%)およびブトルファノール(Imax:83%)と比較して統計学的に有意に低値
であり、ブプレノルフィン(Imax:80%)と同程度であった。EC50の比較では、DPDPE(0.186 nmol/L)
< ブプレノルフィン(2.40 nmol/L)< ブトルファノール(4.88 nmol/L)< TRK-820(21.3 nmol/L)
であり、TRK-820 は最も高濃度であった(図 2.6.2-1、表 2.6.2-4)。なお、モルヒネは 10000 nmol/L
においても最大反応を示さなかったため、試験では Imaxおよび EC50は算出しなかったが、
10000 nmol/L での反応率が約 80%であることから、他の被験物質と同等の方法により、EC50を算
出した結果、394 nmol/L となった。
オピオイド κ、μ および δ 受容体に対する作動性について、各薬剤の EC50 の比(κ:μ:δ)は、
モルヒネで 1:0.1:1.0、ブプレノルフィンで 1:0.4:0.6、ブトルファノールで 1:4.4:6.5 およ
び TRK-820 で 1:203:2610 であることから、TRK-820 は、モルヒネ、ブプレノルフィンおよび
ブトルファノールと明確にプロファイルが異なり、オピオイド κ 受容体に対して選択的で非常に
強い作動性を有することが示唆された。
- 25 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 18
図 2.6.2-1. TRK-820 および既存オピオイド受容体作動薬のヒトオピオイド受容体作動性
フォルスコリン刺激による cAMP 産生に対する抑制作用を示す。 縦軸: フォルスコリン単独処置による cAMP 産生量を 100%としたときの cAMP 産生率(%) 各点は平均値 ± 標準誤差(n=5)を示す。 各受容体の完全作動薬として U-69593(オピオイド κ受容体)、DAMGO(オピオイド μ受容体)、DPDPE(オピ
オイド δ受容体)を使用した。
10 -14 10-13 10-12 10 -11 10-10 10 -9 10 -8 10-7 10 -6 10-50
20
40
60
80
100
120
10 -14 10-13 10-12 10-11 10 -10 10 -9 10-8 10-7 10 -6 10-50
20
40
60
80
100
120
10 -14 10-13 10 -12 10-11 10-10 10 -9 10 -8 10-7 10-6 10-50
20
40
60
80
100
120
cAM
P
産生率(
%)
cAM
P
産生率(
%)
cAM
P
産生率(
%)
A. オピオイド受容体 B. オピオイド受容体
C. オピオイド受容体
濃度 (mol/L) 濃度 (mol/L)
濃度 (mol/L)
:各受容体の完全作動薬
:TRK-820
:モルヒネ
:ブプレノルフィン
:ブトルファノール
- 26 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 19
表 2.6.2-4. TRK-820 および既存オピオイド受容体作動薬のヒトオピオイド受容体作動性
被験物質 オピオイド κ 受容体 オピオイド μ 受容体 オピオイド δ 受容体
EC50 (nmol/L)
Imax (%)
EC50 (nmol/L)
Imax (%)
EC50 (nmol/L)
Imax (%)
TRK-820 0.00816 ± 0.00138 91.3 ± 0.5 1.66 ± 0.09 53.2 ± 1.3 a,b,d 21.3 ± 1.0 77.9 ± 1.6
d,e
モルヒネ 391 ± 33 80.4 ± 0.7 35.7 ± 2.6 75.6 ± 0.5 N.C. (80.5 ± 0.7) f
ブプレノルフィン 4.13 ± 0.24 47.7 ± 1.7 1.59 ± 0.26 56.1 ± 1.1 a,b 2.40 ± 0.19 79.9 ± 1.5 e ブトルファノール 0.752 ± 0.050 84.5 ± 0.8 3.34 ± 0.23 46.5 ± 1.0 a,b,c 4.88 ± 0.41 83.3 ± 1.0 各受容体の完全作動薬 g 0.642 ± 0.022 91.2 ± 0.3 5.63 ± 0.31 77.2 ± 0.8 0.186 ± 0.045 87.3 ± 0.6
数値は平均値 ± 標準誤差(n=5)を示す。 N.C.: 最高濃度において最大反応に達していなかったため、算出しなかった。 a: p<0.05 vs. DAMGO(Tukey 型多重比較) b: p<0.05 vs. モルヒネ(Tukey 型多重比較) c: p<0.05 vs. ブプレノルフィン(Tukey 型多重比較) d: p<0.05 vs. ブトルファノール(Tukey 型多重比較) e: p<0.05 vs. DPDPE(Tukey 型多重比較) f: 最高濃度において最大反応に達していなかったため、参考値として最高濃度での反応率を示した。 g: U-69593(オピオイド κ 受容体)、DAMGO(オピオイド μ 受容体)、DPDPE(オピオイド δ 受容体)
2.6.2.2.2 引っ掻き行動抑制作用(in vivo)
TRK-820 の止痒作用をマウスのヒスタミン皮内投与誘発引っ掻き行動、サブスタンス P 皮内投
与誘発引っ掻き行動、デオキシコール酸皮内投与誘発引っ掻き行動、モルヒネ大槽内投与誘発引
っ掻き行動および自然発症アトピー性皮膚炎マウスの引っ掻き行動を指標に評価した。なお、
TRK-820 の投与は、臨床投与経路である経口にて実施した。ただし、モルヒネ大槽内投与モデル
においてのみ TRK-820 は皮下投与した。
(1) ヒスタミン誘発引っ掻き行動に対する作用(2.6.3.2 (2)、4.2.1.1-5: 試験、
4.2.1.1-6: 試験、4.2.1.1-7: 試験)
a) 方法
ICR 系雄性マウスの吻側背部皮内に、ヒスタミン(10 μg/50 μL/site)を投与することにより誘発
される引っ掻き行動を、ヒスタミン投与直後から 30 分後まで測定した。ヒスタミン皮内投与の
30 分前に TRK-820(3、10、30 および 100 μg/kg)を経口投与し、引っ掻き行動の抑制作用を指標
に止痒作用を評価した。また、ケトチフェン(0.3、3 および 30 mg/kg)またはクロルフェニラミ
ン(3、10 および 30 mg/kg)をヒスタミン皮内投与の 60 分前に経口投与し、引っ掻き行動抑制作
用を観察し、TRK-820 の作用と比較した。
- 27 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 20
b) 成績
TRK-820 は、用量依存的にヒスタミン誘発引っ掻き行動回数を減少させた。30 および 100 μg/kg
投与群では、溶媒対照群と比較して統計学的に有意な作用が認められた(図 2.6.2-2)。TRK-820
の ED50は 7.30 μg/kg(95%信頼区間:4.22~12.6 μg/kg)であった。比較対照薬のケトチフェンお
よびクロルフェニラミンも用量依存的にヒスタミン誘発引っ掻き行動回数を減少させ(図
2.6.2-2)、ED50はそれぞれ 3.35 mg/kg(95%信頼区間:0.554~20.3 mg/kg)および 8.50 mg/kg(95%
信頼区間:1.73~25.5 mg/kg)であった。以上のように、TRK-820 は抗ヒスタミン薬が有効な痒み
に対して止痒作用を有している可能性が示唆された。
- 28 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 21
図 2.6.2-2. ヒスタミン誘発引っ掻き行動に対する作用
被験物質投与から起痒剤投与までの時間 TRK-820(経口投与): 30 分、ケトチフェン(経口投与): 60 分 クロルフェニラミン(経口投与): 60 分 起痒剤なし: PBS(起痒剤の溶媒)を 50 μL/site の容量で皮内投与 被験物質非投与群: 蒸留水(溶媒)を 10 mL/kg の容量で経口投与(TRK-820、クロルフェニラミン) 10 vol% DMSO 水溶液(溶媒)を 10 mL/kg の容量で経口投与(ケトチフェン) 縦軸: 30 分間の引っ掻き行動回数(平均値 ± 標準誤差) n=8: TRK-820(30 μg/kg 投与群のみ n=7)、n=12: ケトチフェン、 n=9: クロルフェニラミン #p<0.05、##p<0.01(TRK-820: Welch 検定、ケトチフェンおよびクロルフェニラミン: t 検定) *p<0.05、**p<0.01 vs. 起痒剤あり・被験物質非投与群(Dunnett 型多重比較)
0 0 0.3 3 300
25
50
75
100
0 0 3 10 30 1000
50
100
150
200
0 0 3 10 300
50
100
150
A. TRK-820 B. ケトチフェン
C. クロルフェニラミン
:起痒剤なし
:起痒剤あり (ヒスタミン10 µg/site, i.d.)
引っ掻き行動回数/30分
引っ掻き行動回数/30分
引っ掻き行動回数/30分
(µg/kg, p.o.) (mg/kg, p.o.)
(mg/kg, p.o.)
##
*
**
#
*
##
- 29 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 22
(2) サブスタンスP誘発引っ掻き行動に対する作用(2.6.3.2 (2)、4.2.1.1-8: 試験、
4.2.1.1-9: 試験、4.2.1.1-10: 試験、4.2.1.1-11: 試験)
a) 方法
ICR 系雄性マウスの吻側背部皮内に、サブスタンス P(250 nmol/50 μL/site)を投与することに
より誘発される引っ掻き行動を、サブスタンス P 投与直後から 30 分後まで測定した。サブスタン
ス P 皮内投与の 30 分前に TRK-820(3、10、30 および 100 μg/kg)を経口投与し、引っ掻き行動
の抑制作用を指標に止痒作用を評価した。また、ケトチフェン(0.1、1、10 および 100 mg/kg)ま
たはクロルフェニラミン(1、3、10 および 30 mg/kg)をサブスタンス P 皮内投与の 60 分前に経
口投与、オキサトミド(0.1、1、10 および 100 mg/kg)をサブスタンス P 皮内投与の 120 分前に経
口投与して引っ掻き行動抑制作用を観察し、TRK-820 の作用と比較した。
b) 成績
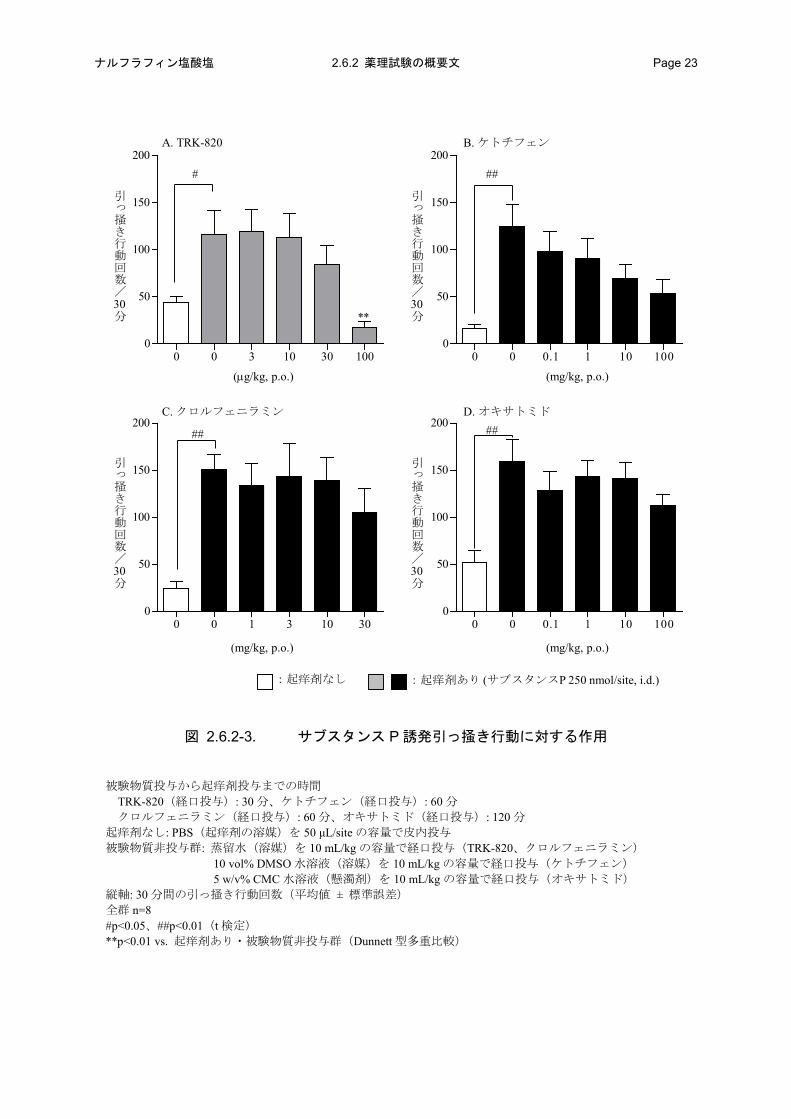
TRK-820 は、用量依存的にサブスタンス P 誘発引っ掻き行動回数を減少させた。100 μg/kg 投与
群では溶媒対照群と比較して統計学的に有意な作用が認められた(図 2.6.2-3)。TRK-820 の ED50
は 19.6 μg/kg(95%信頼区間:9.59~40.0 μg/kg)であった。比較対照薬のケトチフェンでは用量依
存的にサブスタンス P 誘発引っ掻き行動回数を減少させたが、試験で使用した最高用量の
100 mg/kg 投与群においても溶媒対照群と比較して抑制率は 66%であり、有意差も認められなかっ
た(図 2.6.2-3)。なお、ケトチフェンの ED50は 9.61 mg/kg(95%信頼区間:0.541~171 mg/kg)で
あった。また、クロルフェニラミンおよびオキサトミドはサブスタンス P による引っ掻き行動を
抑制しなかった(図 2.6.2-3)。以上のように、TRK-820 は抗ヒスタミン薬が十分な効果を示さな
い痒みに対して止痒作用を有する可能性が示唆された。
- 30 -
ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 23
図 2.6.2-3. サブスタンス P 誘発引っ掻き行動に対する作用
被験物質投与から起痒剤投与までの時間 TRK-820(経口投与): 30 分、ケトチフェン(経口投与): 60 分 クロルフェニラミン(経口投与): 60 分、オキサトミド(経口投与): 120 分 起痒剤なし: PBS(起痒剤の溶媒)を 50 μL/site の容量で皮内投与 被験物質非投与群: 蒸留水(溶媒)を 10 mL/kg の容量で経口投与(TRK-820、クロルフェニラミン) 10 vol% DMSO 水溶液(溶媒)を 10 mL/kg の容量で経口投与(ケトチフェン) 5 w/v% CMC 水溶液(懸濁剤)を 10 mL/kg の容量で経口投与(オキサトミド) 縦軸: 30 分間の引っ掻き行動回数(平均値 ± 標準誤差) 全群 n=8 #p<0.05、##p<0.01(t 検定) **p<0.01 vs. 起痒剤あり・被験物質非投与群(Dunnett 型多重比較)
0 0 0.1 1 10 1000
50
100
150
200
0 0 0.1 1 10 1000
50
100
150
200
0 0 3 10 30 1000
50
100
150
200
0 0 1 3 10 300
50
100
150
200
A. TRK-820 B. ケトチフェン
C. クロルフェニラミン
:起痒剤なし :起痒剤あり (サブスタンスP 250 nmol/site, i.d.)
引っ掻き行動回数/30分
引っ掻き行動回数/30分
引っ掻き行動回数/30分
(µg/kg, p.o.) (mg/kg, p.o.)
(mg/kg, p.o.)
#
**
##
##
引っ掻き行動回数/30分
##D. オキサトミド
(mg/kg, p.o.)
- 31 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 24
(3) デオキシコール酸誘発引っ掻き行動に対する作用(2.6.3.2 (2)、4.2.1.1-12: 試験)
a) 方法
ICR 系雄性マウスの吻側背部皮内に、胆汁酸の構成成分の一つであるデオキシコール酸
(100 μg/50 μL/site)を投与することにより誘発される引っ掻き行動を、デオキシコール酸投与直
後から 30 分後まで測定した。デオキシコール酸皮内投与の 30 分前に TRK-820(3、10、30 およ
び 100 μg/kg)を経口投与し、引っ掻き行動の抑制作用を指標に胆汁うっ滞性の痒みに対する止痒
作用を評価した。また、ケトチフェン(1、3、10 および 30 mg/kg)の経口投与またはオピオイド
μ 受容体拮抗薬ナルトレキソン(0.3、1、3 および 10 mg/kg)の皮下投与をデオキシコール酸皮内
投与のそれぞれ 60 または 30 分前に行い、引っ掻き行動抑制作用を観察し、TRK-820 の作用と比
較した。
b) 成績
TRK-820 は、用量依存的にデオキシコール酸誘発引っ掻き行動回数を減少させ、30 および
100 μg/kg 投与群では溶媒対照群と比較して統計学的に有意な作用が認められた(図 2.6.2-4)。
TRK-820 の ED50 は 7.62 μg/kg(95%信頼区間:3.91~12.0 μg/kg)であった。比較対照薬のケトチ
フェンはデオキシコール酸による引っ掻き行動を抑制しなかった(図 2.6.2-4)。なお、臨床にお
いて胆汁うっ滞性の痒みに有効であることが報告されているオピオイド μ 受容体拮抗薬ナルトレ
キソン 7)は、デオキシコール酸誘発の引っ掻き行動を、試験で使用した最低用量の 0.3 mg/kg にお
いて有意に抑制した。しかしながら、ナルトレキソンは用量を増加させても 60~70%の引っ掻き
行動抑制作用しか示さず、作用の頭打ちが認められた(図 2.6.2-4)。なお、ナルトレキソンの ED50
は、0.173 mg/kg(95%信頼区間算出不能)であった。以上のように、TRK-820 は抗ヒスタミン薬
が無効な胆汁うっ滞性の痒みに対して止痒作用を有する可能性が示唆された。
- 32 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 25
図 2.6.2-4. デオキシコール酸誘発引っ掻き行動に対する作用
被験物質投与から起痒剤投与までの時間 TRK-820(経口投与): 30 分、ケトチフェン(経口投与): 60 分 ナルトレキソン(皮下投与): 30 分 起痒剤なし: PBS(起痒剤の溶媒)を 50 μL/site の容量で皮内投与 被験物質非投与群: 蒸留水(溶媒)を 10 mL/kg の容量で経口投与(TRK-820、ケトチフェン) 生理食塩液(溶媒)を 10 mL/kg の容量で皮下投与(ナルトレキソン) 縦軸: 30 分間の引っ掻き行動回数(平均値 ± 標準誤差) 全群 n=8 ##p<0.01(TRK-820 およびナルトレキソン: Welch 検定、ケトチフェン: t 検定) *p<0.05、**p<0.01 vs. 起痒剤あり・被験物質非投与群(Dunnett 型多重比較)
0 0 1 3 10 300
50
100
150
200
250
0 0 3 10 30 1000
50
100
150
200
250
0 0 0.3 1 3 100
50
100
150
200
250
A. TRK-820 B. ケトチフェン
C. ナルトレキソン
:起痒剤なし
:起痒剤あり(デオキシコール酸 100 µg/site, i.d.)
引っ掻き行動回数/30分
引っ掻き行動回数/30分
引っ掻き行動回数/30分
(µg/kg, p.o.) (mg/kg, p.o.)
(mg/kg, s.c.)
##
*
**
##
*
##
*
* **
- 33 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 26
(4) モルヒネ大槽内投与誘発引っ掻き行動に対する作用(2.6.3.2 (2)、4.2.1.1-13: 試
験、4.2.1.1-14: 試験)
a) 方法
ddY 系雄性マウスの大槽内(小脳延髄槽)に、モルヒネ(0.3 nmol/5 μL/site)を投与することに
より誘発される引っ掻き行動を、モルヒネ投与直後から 60 分後まで測定した。モルヒネ大槽内投
与の 30 分前に TRK-820(1.25、2.5、5 および 10 μg/kg)を皮下投与し、引っ掻き行動の抑制作用
を指標に止痒作用を評価した。また、ケトチフェン(0.01、0.1、1 および 10 mg/kg)をモルヒネ
大槽内投与の 30 分前に腹腔内投与して引っ掻き行動抑制作用を観察し、TRK-820 の作用と比較し
た。なお、本モデルの引っ掻き行動は、オピオイド μ 受容体拮抗薬ナロキソンにより抑制される
ことから、中枢のオピオイド μ 受容体の活性化に基づく行動であることが報告されている 9)。
b) 成績
TRK-820 は、用量依存的にモルヒネ誘発引っ掻き行動回数を減少させた。5 および 10 μg/kg 投
与群では溶媒対照群と比較して統計学的に有意な作用が認められた(図 2.6.2-5)。TRK-820 の ED50
は 2.34 μg/kg(95%信頼区間:1.28~3.34 μg/kg)であった。比較対照薬のケトチフェンは、低用量
側(0.01~0.1 mg/kg)では溶媒対照群と比較して引っ掻き行動が増加したが統計学的に有意な差
は認められなかった。一方、ケトチフェン 1 mg/kg 投与群の引っ掻き行動回数は、溶媒対照群と
同程度であった。ケトチフェン 10 mg/kg 投与群では、引っ掻き行動回数は減少したが、溶媒対照
群と比較して統計学的に有意な差は認められなかった(図 2.6.2-5)。したがって、TRK-820 は抗
ヒスタミン薬が無効な中枢神経系のオピオイド μ 受容体の活性化に伴う痒みに対しても止痒作用
を有する可能性が示唆された。
- 34 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 27
図 2.6.2-5. モルヒネ大槽内投与誘発引っ掻き行動に対する作用
被験物質投与から起痒剤投与までの時間 TRK-820(皮下投与): 30 分、ケトチフェン(腹腔内投与): 30 分 起痒剤なし: 生理食塩液(溶媒)を 5 μL/site の容量で大槽内投与 被験物質非投与群: 生理食塩液(溶媒)を 10 mL/kg の容量で皮下(TRK-820)もしくは腹腔内(ケトチフェン)
投与 縦軸: 60 分間の引っ掻き行動回数(平均値 ± 標準誤差) カラム内の数字: 各群の例数 ##p<0.01(Welch 検定) *p<0.05、**p<0.01 vs. 起痒剤あり・被験物質非投与群(Dunnett 型多重比較)
(5) 自然発症アトピー性皮膚炎モデルの引っ掻き行動に対する作用(2.6.3.2 (2)、4.2.1.1-15:試験)
a) 方法
NC/Nga 系雄性マウスをコンベンショナル環境下で飼育し、皮膚炎を自然発症させた。皮膚炎発
症マウスに認められる引っ掻き行動を 90 分間測定した。測定開始の 30 分前に TRK-820(10、30
および 100 μg/kg)を経口投与し、引っ掻き行動の抑制作用を指標にアトピー性皮膚炎に伴う痒み
に対する止痒作用を評価した。なお、本モデルの引っ掻き行動はクロルフェニラミンもしくはケ
トチフェンで抑制できないことが報告されている 10-12)。
b) 成績
バリアシステムの環境下で飼育した SPF NC/Nga 系雄性マウスと比較して、コンベンショナル
環境下で飼育し、皮膚炎を発症させたマウスでは引っ掻き行動が増加し、統計学的に有意な差が
認められた。TRK-820 は、用量の増加に伴って皮膚炎発症マウスの引っ掻き行動回数を減少させ
0 0 1.25 2.5 5 100
20
40
60
80
100
0 0 0.01 0.1 1 100
50
100
150
(mg/kg, i.p.)
A. TRK-820 B. ケトチフェン
:起痒剤なし :起痒剤あり (モルヒネ 0.3 nmol/site, i.c.)
引っ掻き行動回数/60分
引っ掻き行動回数/60分
(µg/kg, s.c.)
##
**
*
##
**
**15 12 11 11 12 14 14 10 13 11 10 12
- 35 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 28
た。100 μg/kg 投与群では溶媒対照群と比較して統計学的に有意な作用が認められた(図 2.6.2-6)。
TRK-820 の ED50 は 46.1 μg/kg(95%信頼区間:25.7~125 μg/kg)であった。したがって、TRK-820
は抗ヒスタミン薬の効果が不十分であるといわれているアトピー性皮膚炎に伴う痒みに対して止
痒作用を有する可能性が示唆された。
図 2.6.2-6. 皮膚炎自然発症 NC/Nga 系マウスの引っ掻き行動に対する作用
TRK-820 経口投与から測定開始までの時間: 30 分 被験物質非投与群: 蒸留水(溶媒)を 10 mL/kg の容量で経口投与 縦軸: 90 分間の引っ掻き行動回数(平均値 ± 標準誤差) SPF NC マウス: バリアシステムの環境下で飼育した NC/Nga 系マウス コンベンショナル NC マウス: コンベンショナル環境下で飼育した NC/Nga 系マウス n=8(ただし、SPF NC マウスおよび 10 μg/kg 投与群は n=7) ##p<0.01(Welch 検定) **p<0.01 vs. コンベンショナル NC マウス・被験物質非投与群(Dunnett 型多重比較)
2.6.2.2.3 引っ掻き行動抑制作用の持続時間(in vivo)(2.6.3.2 (2)、4.2.1.1-16:試験)
a) 方法
ddY 系雄性マウスの吻側背部皮内に、サブスタンス P(250 nmol/50 μL/site)を投与することに
より誘発される引っ掻き行動を、サブスタンス P 投与直後から 30 分後まで測定した。サブスタン
ス P 皮内投与の 0.5、2、4、6 または 8 時間前に TRK-820(100 μg/kg)を経口投与し、引っ掻き行
動の抑制作用を指標に止痒作用を評価した。
0 0 10 30 1000
100
200
300
400
500
:SPF NC マウス
:コンベンショナル NC マウス
引っ掻き行動回数/90分
TRK-820 (µg/kg, p.o.)
##
**
- 36 -
ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 29
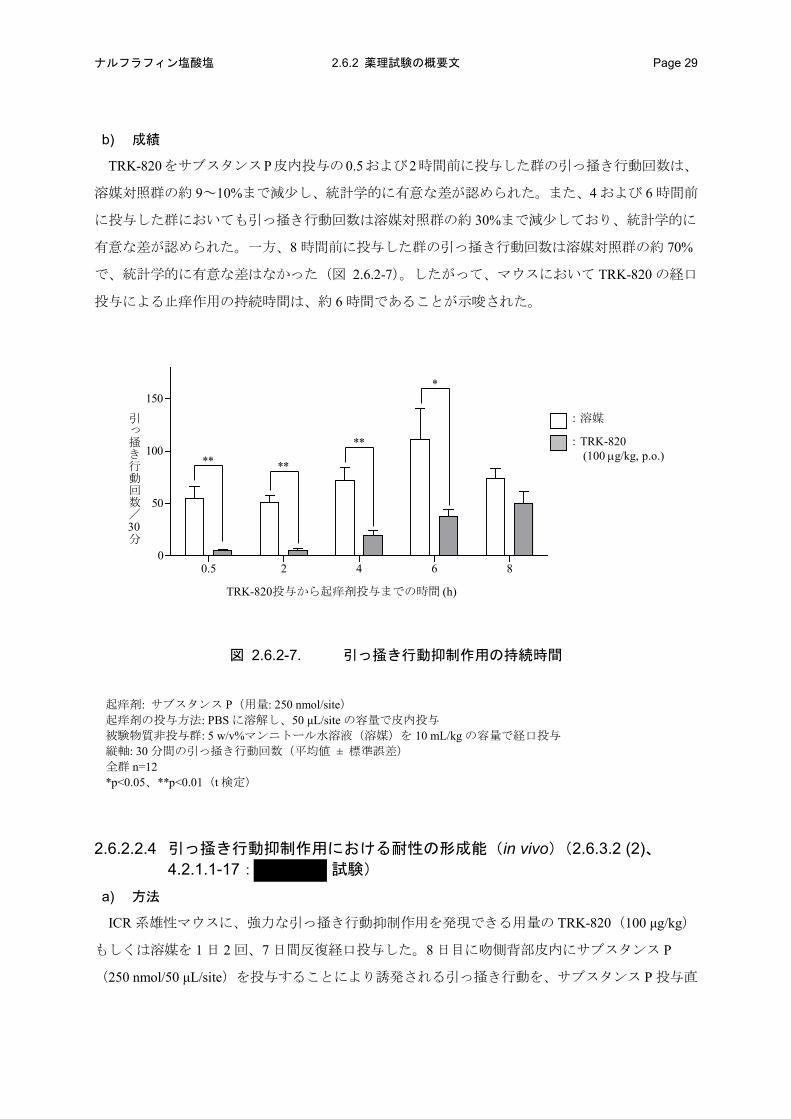
b) 成績
TRK-820をサブスタンスP皮内投与の0.5および2時間前に投与した群の引っ掻き行動回数は、
溶媒対照群の約 9~10%まで減少し、統計学的に有意な差が認められた。また、4 および 6 時間前
に投与した群においても引っ掻き行動回数は溶媒対照群の約 30%まで減少しており、統計学的に
有意な差が認められた。一方、8 時間前に投与した群の引っ掻き行動回数は溶媒対照群の約 70%
で、統計学的に有意な差はなかった(図 2.6.2-7)。したがって、マウスにおいて TRK-820 の経口
投与による止痒作用の持続時間は、約 6 時間であることが示唆された。
図 2.6.2-7. 引っ掻き行動抑制作用の持続時間
起痒剤: サブスタンス P(用量: 250 nmol/site) 起痒剤の投与方法: PBS に溶解し、50 μL/site の容量で皮内投与 被験物質非投与群: 5 w/v%マンニトール水溶液(溶媒)を 10 mL/kg の容量で経口投与 縦軸: 30 分間の引っ掻き行動回数(平均値 ± 標準誤差) 全群 n=12 *p<0.05、**p<0.01(t 検定)
2.6.2.2.4 引っ掻き行動抑制作用における耐性の形成能(in vivo)(2.6.3.2 (2)、4.2.1.1-17: 試験)
a) 方法
ICR 系雄性マウスに、強力な引っ掻き行動抑制作用を発現できる用量の TRK-820(100 μg/kg)
もしくは溶媒を 1 日 2 回、7 日間反復経口投与した。8 日目に吻側背部皮内にサブスタンス P
(250 nmol/50 μL/site)を投与することにより誘発される引っ掻き行動を、サブスタンス P 投与直
0.5 2 4 6 80
50
100
150
:溶媒
:TRK-820(100 µg/kg, p.o.)
引っ掻き行動回数/30分
** **
**
*
TRK-820投与から起痒剤投与までの時間 (h)
- 37 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 30
後から 30 分後まで測定した。サブスタンス P 皮内投与の 30 分前に TRK-820(25~200 μg/kg)を
経口投与し、引っ掻き行動の抑制作用をTRK-820反復投与群と溶媒反復投与群との間で比較した。
b) 成績
溶媒反復投与群ではTRK-820 の 25 μg/kg 以上で統計学的に有意な引っ掻き行動抑制作用が発現
した(図 2.6.2-8)。一方、TRK-820 反復投与群では 100 μg/kg 以上で統計学的に有意な引っ掻き行
動抑制作用が認められた(図 2.6.2-8)。したがって、TRK-820 の反復経口投与により引っ掻き行
動抑制作用は減弱することが示唆された。モルヒネの鎮痛作用に対する耐性形成に関する文献で
は、鎮痛作用が認められる用量のモルヒネを 1 日 1 回、6 日間 13)もしくは 1 日 2 回、9 日間 14)反
復投与すると鎮痛作用がほぼ完全に消失することが報告されている。一方、TRK-820 の止痒作用
が認められる用量の反復投与によって、同用量の TRK-820 の引っ掻き行動抑制作用は消失してい
ないこと、また、溶媒反復投与群および TRK-820 反復投与群の ED50 はそれぞれ 30.4 μg/kg(95%
信頼区間:20.5~39.5 μg/kg)および 56.0 μg/kg(95%信頼区間:40.7~72.7 μg/kg)であり、TRK-820
反復投与によって ED50は 1.8 倍に増加したのみであったことから、TRK-820 が止痒作用において
強い耐性を形成する可能性は低いと考えられた。
- 38 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 31
図 2.6.2-8. 引っ掻き行動抑制作用における耐性の形成能
A: 蒸留水(溶媒)を 10 mL/kg の容量で 1 日 2 回、7 日間反復経口投与 B: TRK-820 を 100 μg/kg の用量で 1 日 2 回、7 日間反復経口投与 反復経口投与終了の翌日に引っ掻き行動を評価 TRK-820 経口投与から起痒剤投与までの時間: 30 分 起痒剤なし: PBS(起痒剤の溶媒)を 50 μL/site の容量で皮内投与 被験物質非投与群: 蒸留水を 10 mL/kg の容量で経口投与 縦軸: 30 分間の引っ掻き行動回数(平均値 ± 標準誤差) n=8(ただし、起痒剤なし・被験物質非投与群は n=7) ##p<0.01(溶媒反復経口投与: t 検定、TRK-820 反復経口投与: Welch 検定) **p<0.01 vs. 起痒剤あり・被験物質非投与群(Dunnett 型多重比較)
2.6.2.2.5 作用機序
(1) 炎症性メディエーター遊離もしくは分泌およびNOS活性に対する作用(in vitro)(2.6.3.2 (3)、4.2.1.1-18: 試験)
a) 方法
サブスタンス P(10 μmol/L)またはコンパウンド 48/80(0.15 μg/mL)刺激によるラット肥満細
胞からのヒスタミン遊離、LPS(30 μg/mL)刺激による THP-1 細胞からの TNF-α、IL-1β および IL-6
分泌、A23187(8 μmol/L)刺激による HL-60 細胞からの PGE2 分泌、A23187(10 μmol/L)刺激に
よるラット肥満細胞からの PGD2 分泌、[3H]アルギニンを基質とした RAW264-7 細胞の誘導型 NOS
活性および HUVEC 細胞の構成型 NOS 活性に対する TRK-820(1 および 1000 nmol/L)の阻害作
用を評価した。
0 0 25 50 100 2000
50
100
150
200
250
0 0 25 50 100 2000
50
100
150
200
250
300
350
A. 溶媒反復経口投与群 B. TRK-820反復経口投与群
:起痒剤なし :起痒剤あり (サブスタンス P 250 nmol/site, i.d.)
引っ掻き行動回数/30分
引っ掻き行動回数/30分
TRK-820 (µg/kg, p.o.)
##
**
##
**
**
TRK-820 (µg/kg, p.o.)
**
**
**
- 39 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 32
b) 成績
TRK-820の1 nmol/Lは、サブスタンスP刺激によるラット肥満細胞からのヒスタミン遊離を12%
阻害し、LPS 刺激による THP-1 細胞からの IL-1β および IL-6 分泌をそれぞれ 19 および 11%阻害
した。しかしながら、高濃度の TRK-820 の 1000 nmol/L では、すべての炎症性メディエーター遊
離に対して阻害作用は認められなかった(表 2.6.2-5)。また、TRK-820 の 1000 nmol/L は、誘導型
および構成型 NOS 活性に対して阻害作用を示さなかった(表 2.6.2-5)。したがって、TRK-820 の
止痒作用は、炎症性メディエーター遊離もしくは分泌および NOS 活性の阻害作用には起因しない
ものと考えられた。
表 2.6.2-5. 炎症性メディエーター遊離もしくは分泌および NOS 活性に対する作用
評価項目 試験系 TRK-820 による
阻害率 (%) 対照化合物IC50
(× 1000 nmol/L) 細胞 刺激剤/基質 1 nmol/La
1000 nmol/La
ヒスタミン遊離 ラット肥満細胞 サブスタンス P (10 μmol/L) 12 < 10 N.E.
ヒスタミン遊離 ラット肥満細胞 コンパウンド 48/80 (0.15 μg/mL) < 10 < 10
Sodium cromoglicate
92
TNF-α 分泌 THP-1 細胞 LPS (30 μg/mL) < 10 < 10 Staurosporine 0.035
IL-1β 分泌 THP-1 細胞 LPS (30 μg/mL) 19 < 10 Dexamethasone 0.0021
IL-6 分泌 THP-1 細胞 LPS (30 μg/mL) 11 < 10 Dexamethasone 0.00088
PGE2分泌 HL-60 細胞 A23187 (8 μmol/L) < 10 < 10 Indomethacin 0.0014
PGD2 分泌 ラット肥満細胞 A23187 (10 μmol/L) < 10 < 10 Indomethacin 0.11
誘導型 NOS 活性 RAW264-7 細胞 [3H]アルギニン (28 nmol/L) N.E. < 10 L-NMMAb
1.2
構成型 NOS 活性 HUVEC 細胞 [3H]アルギニン (56 nmol/L) N.E. < 10 L-NMMAb
0.12 TRK-820 による阻害率(%)は 1 回の実験(n=2)から算出した。 N.E.: 実施せず、A23187: カルシウムイオノフォア a: 試験に用いた TRK-820 の濃度 b: NG-monomethyl-L-arginine
- 40 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 33
(2) オピオイド受容体以外の受容体、イオンチャネルまたはトランスポーターに対する作用
(in vitro)(2.6.3.2 (3)、4.2.1.1-18: 試験、4.2.1.1-19: 試験、4.2.1.1-20:試験、4.2.1.1-21: 試験)
特異的リガンドの結合に対する TRK-820 の阻害作用を指標として、各受容体、イオンチャネル
またはトランスポーターに対する結合性を評価した。
a) 方法
各受容体、イオンチャネルまたはトランスポーターへの結合試験に用いたリガンドと標本を示
した(表 2.6.2-6)。
- 41 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 34
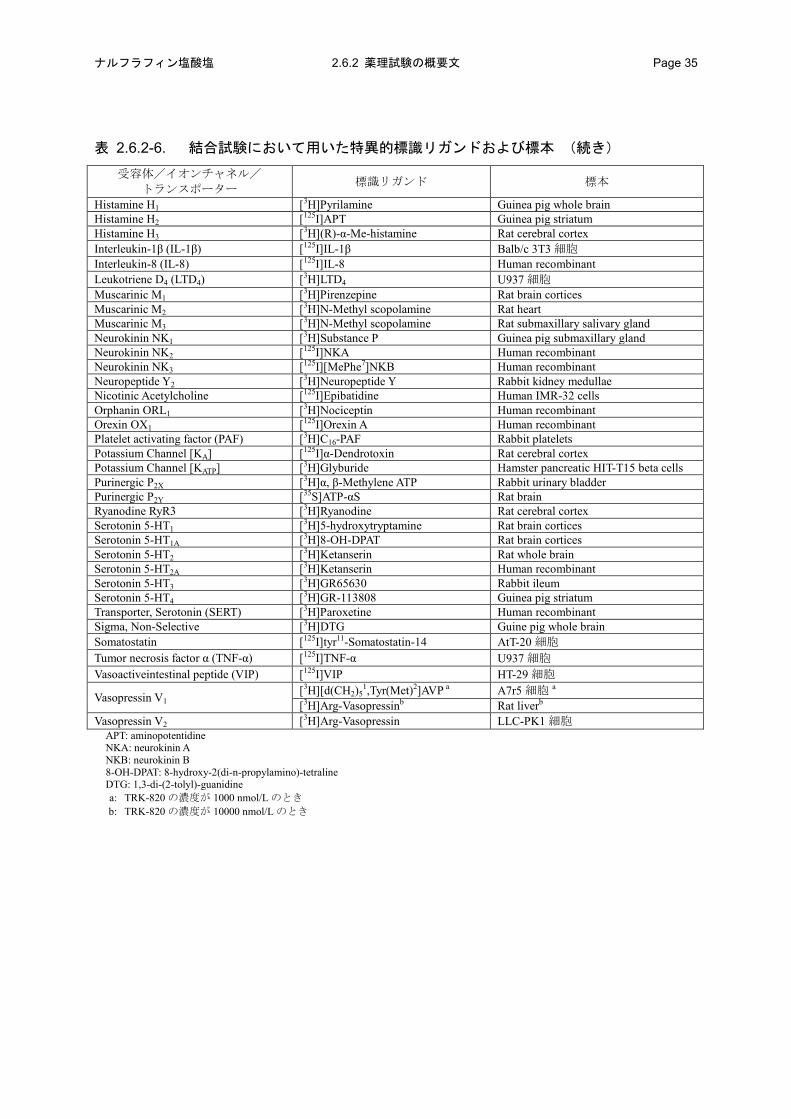
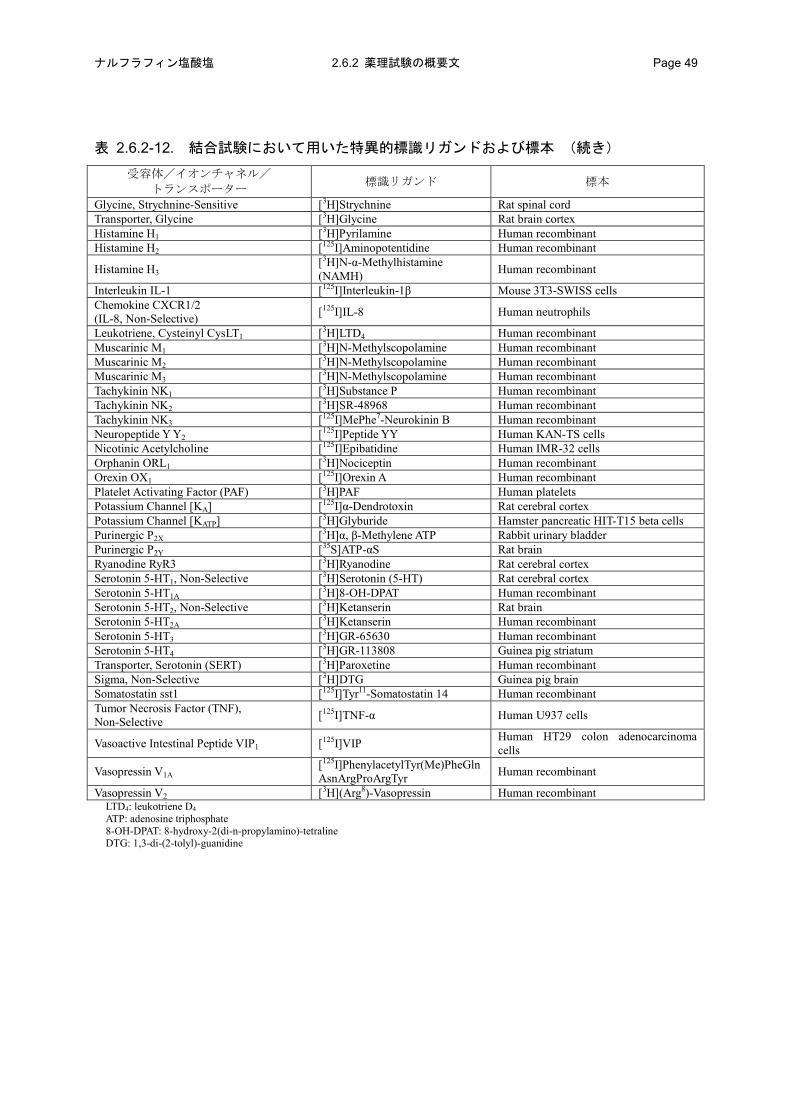
表 2.6.2-6. 結合試験において用いた特異的標識リガンドおよび標本
受容体/イオンチャネル/ トランスポーター 標識リガンド 標本
Adenosine A1 [3H]Cyclopentyl-1,3-dipropylxanthine Rat whole brain Adenosine A2A [3H]CGS-21680 Rat striata Transporter, Adenosine [3H]Nitrobenzylthioinosine Guinea pig cerebral cortex Adrenergic α1, Non-Selective [3H]Prazosin Rat whole brain Adrenergic α2, Non-Selective [3H]Rauwolscine Rat brain cortices Adrenergic β1 [125I]Cyanopindolol Human recombinant Adrenergic β2 [3H]CGP-12177 Human recombinant Transporter, Norepinephrine (NET) [125I]RTI-55 Human recombinant Angiotensin II [3H]Angiotensin II Rabbit adrenal gland Atrial natriuretic peptide (ANP) [125I]ANP Guinea pig cerebellum Bombesin [125I]Tyr4-Bombesin Rat whole brain Bradykinin B2 [3H]Bradykinin Human recombinant Calcitonin gene-related peptide (CGRP) [125I]hCGRPα SK-N-MC 細胞
Calcium channel Type L, Dihydropyridine site [3H]Nitrendipine Rat brain cortices
Calcium Channel N-Type [125I]ω-Conotoxin GVIA Rat frontal brain Cannabinoid CB1 [3H]SR141716A Human recombinant CC Chemokine receptor CCR1 [125I]MIP-1α Human recombinant CC Chemokine receptor CCR2 [125I]MCP-1 Human recombinant Cholecystokinin A [3H]L-364718 Guinea pig pancreas Cholecystokinin B [3H]Cholecystokinin-8 Mouse brain Transporter, Choline [3H]Hemicholinium-3 Rat brain striatum Corticotropin Releasing Factor CRF1 [125I](Tyr0)-CRF (ovine) Human recombinant Dopamine D1 [3H]SCH-23390 Rat striata Dopamine D2 [3H]Spiperone Rat striata Dopamine D3 [3H]Spiperone Human recombinant Transporter, Dopamine (DAT) [125I]RTI-55 Human recombinant Transporter, GABA [3H]GABA Rat cerebral cortex GABAA, Agonist Site [3H]Muscimol Rat whole brain GABAA, Flunitrazepam, Central [3H]Flunitrazepam Rat brain (minus cerebellum) GABAA, Chloride Channel, TBPS [35S]TBPS Rat cerebral cortex GABAB, Non-Selective [3H]CGP-54626 Rat brain Glucocorticoid [3H]Dexamethasone Human recombinant Glutamate, Non-Selective [3H]L-Glutamate Rat whole brain Glutamate, AMPA [3H]AMPA Rat brain cortices Glutamate, kainate [3H]Kainate Rat whole brain Glutamate, NMDA, Agonist Site [3H]CGS19755 Rat brain cortices Glutamate, NMDA, Phencyclidine Site [3H]TCP Rat brain cortices Glutamate, Metabotropic, mGlu2 [3H]LY341495 Human recombinant Glutamate, Metabotropic, mGlu5 [3H]Quisqualic acid Human recombinant Glycine, Strychnine-Sensitive [3H]Strychnine Rat spinal cord Transporter, Glycine [3H]Glycine Rat brain cortex
MIP-1α: macrophage inflammatory protein-1α MCP-1: monocyte chemoattractant protein-1 GABA: γ-aminobutyric acid TBPS: t-butylbicyclophosphorothionate AMPA: α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid NMDA: N-methyl-D-aspartic acid TCP: tenocyclidine
- 42 -
ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 35
表 2.6.2-6. 結合試験において用いた特異的標識リガンドおよび標本 (続き)
受容体/イオンチャネル/ トランスポーター 標識リガンド 標本
Histamine H1 [3H]Pyrilamine Guinea pig whole brain Histamine H2 [125I]APT Guinea pig striatum Histamine H3 [3H](R)-α-Me-histamine Rat cerebral cortex Interleukin-1β (IL-1β) [125I]IL-1β Balb/c 3T3 細胞 Interleukin-8 (IL-8) [125I]IL-8 Human recombinant Leukotriene D4 (LTD4) [3H]LTD4 U937 細胞 Muscarinic M1 [3H]Pirenzepine Rat brain cortices Muscarinic M2 [3H]N-Methyl scopolamine Rat heart Muscarinic M3 [3H]N-Methyl scopolamine Rat submaxillary salivary gland Neurokinin NK1 [3H]Substance P Guinea pig submaxillary gland Neurokinin NK2 [125I]NKA Human recombinant Neurokinin NK3 [125I][MePhe7]NKB Human recombinant Neuropeptide Y2 [3H]Neuropeptide Y Rabbit kidney medullae Nicotinic Acetylcholine [125I]Epibatidine Human IMR-32 cells Orphanin ORL1 [3H]Nociceptin Human recombinant Orexin OX1 [125I]Orexin A Human recombinant Platelet activating factor (PAF) [3H]C16-PAF Rabbit platelets Potassium Channel [KA] [125I]α-Dendrotoxin Rat cerebral cortex Potassium Channel [KATP] [3H]Glyburide Hamster pancreatic HIT-T15 beta cells Purinergic P2X [3H]α, β-Methylene ATP Rabbit urinary bladder Purinergic P2Y [35S]ATP-αS Rat brain Ryanodine RyR3 [3H]Ryanodine Rat cerebral cortex Serotonin 5-HT1 [3H]5-hydroxytryptamine Rat brain cortices Serotonin 5-HT1A [3H]8-OH-DPAT Rat brain cortices Serotonin 5-HT2 [3H]Ketanserin Rat whole brain Serotonin 5-HT2A [3H]Ketanserin Human recombinant Serotonin 5-HT3 [3H]GR65630 Rabbit ileum Serotonin 5-HT4 [3H]GR-113808 Guinea pig striatum Transporter, Serotonin (SERT) [3H]Paroxetine Human recombinant Sigma, Non-Selective [3H]DTG Guine pig whole brain Somatostatin [125I]tyr11-Somatostatin-14 AtT-20 細胞 Tumor necrosis factor α (TNF-α) [125I]TNF-α U937 細胞 Vasoactiveintestinal peptide (VIP) [125I]VIP HT-29 細胞
Vasopressin V1 [3H][d(CH2)5
1,Tyr(Met)2]AVP a A7r5 細胞 a [3H]Arg-Vasopressinb Rat liverb
Vasopressin V2 [3H]Arg-Vasopressin LLC-PK1 細胞 APT: aminopotentidine NKA: neurokinin A NKB: neurokinin B 8-OH-DPAT: 8-hydroxy-2(di-n-propylamino)-tetraline DTG: 1,3-di-(2-tolyl)-guanidine a: TRK-820 の濃度が 1000 nmol/L のとき b: TRK-820 の濃度が 10000 nmol/L のとき
- 43 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 36
b) 成績
TRK-820 は、ムスカリン M1 受容体に対する[3H]ピレンゼピンの結合を 1000 および 10000 nmol/L
でそれぞれ 41%および 72%阻害し、その Ki 値は 1700 nmol/L であった。また、TRK-820 の
1000 nmol/L では、CGRP 受容体に対する[125I]hCGRPα の結合を 10%、GABAA受容体 Cl-チャネル
に対する[35S]TBPS の結合を 12%、グルタミン酸 mGluR5 受容体に対する[3H]キスカル酸の結合を
13%、オルファニン ORL1 受容体に対する[3H]ノシセプチンの結合を 47%、オレキシン OX1受容体
に対する[125I]オレキシン A の結合を 25%およびセロトニン 5-HT4 受容体に対する[3H]GR-113808
の結合を 24%阻害した。さらに、TRK-820 の 10000 nmol/L では、アデノシン A2A受容体に対する
[3H]CGS-21680 の結合を 13%、アドレナリン α1受容体に対する[3H]プラゾシンの結合を 27%、ア
ドレナリン α2 受容体に対する[3H]ラウオルシンの結合を 11%、ボンベシン受容体に対する
[125I]Tyr4-ボンベシンの結合を 12%、ドパミン D2受容体に対する[3H]スピペロンの結合を 20%、グ
ルタミン酸 NMDA 受容体(Agonist Site)に対する[3H]CGS19755 の結合を 10%、セロトニン 5-HT2
受容体に対する[3H]ケタンセリンの結合を 12%およびシグマ受容体に対する
[3H]1,3-di-(2-totyl)-guanidine(DTG)の結合を 14%阻害した。したがって、TRK-820 は上記の受容
体に対して低い結合性を示したが、オピオイド κ 受容体に対する結合性(ヒトオピオイド受容体
結合試験における Ki 値:0.244 nmol/L、表 2.6.2-3)と比較すると著しく低かった。その他の受容
体、イオンチャネルおよびトランスポーターに対する各特異的リガンド結合に対する TRK-820 の
1000 または 10000 nmol/L での阻害率は 10%未満であり、結合性はほとんど認められなかった(表
2.6.2-7)。
- 44 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 37
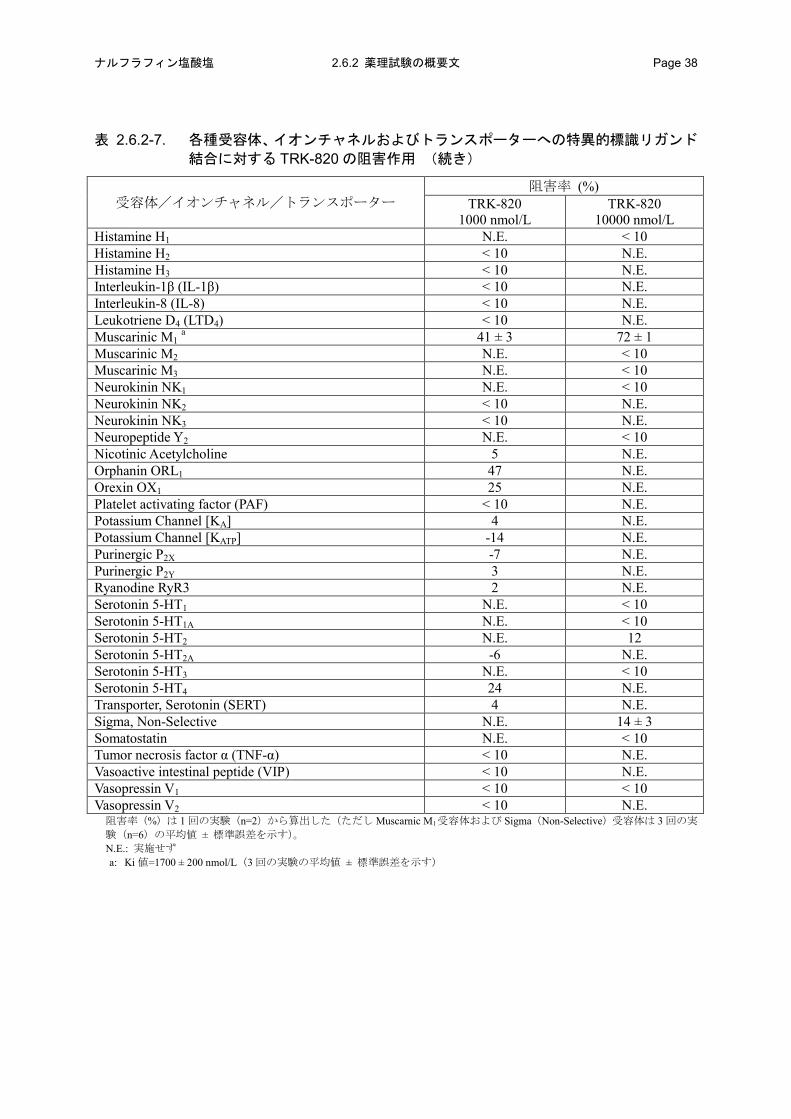
表 2.6.2-7. 各種受容体、イオンチャネルおよびトランスポーターへの特異的標識リガンド
結合に対する TRK-820 の阻害作用
受容体/イオンチャネル/トランスポーター 阻害率 (%)
TRK-820 1000 nmol/L
TRK-820 10000 nmol/L
Adenosine A1 N.E. < 10 Adenosine A2A N.E. 13 Transporter, Adenosine 4 N.E. Adrenergic α1, Non-Selective N.E. 27 Adrenergic α2, Non-Selective N.E. 11 Adrenergic β1 -4 N.E. Adrenergic β2 -3 N.E. Transporter, Norepinephrine (NET) -5 N.E. Angiotensin II N.E. < 10 Atrial natriuretic peptide (ANP) < 10 N.E. Bombesin N.E. 12 Bradykinin B2 4 N.E. Calcitonin gene-related peptide (CGRP) 10 N.E. Calcium channel Type L, Dihydropyridine site N.E. < 10 Calcium Channel N-Type 6 N.E. Cannabinoid CB1 -1 N.E. CC Chemokine receptor CCR1 < 10 N.E. CC Chemokine receptor CCR2 < 10 N.E. Cholecystokinin A N.E. < 10 Cholecystokinin B N.E. < 10 Transporter, Choline -2 N.E. Corticotropin Releasing Factor CRF1 -3 N.E. Dopamine D1 N.E. < 10 Dopamine D2 N.E. 20 Dopamine D3 9 N.E. Transporter, Dopamine (DAT) -11 N.E. Transporter, GABA -3 N.E. GABAA, Agonist Site N.E. < 10 GABAA, Flunitrazepam, Central -1 N.E. GABAA, Chloride Channel, TBPS 12 N.E. GABAB, Non-Selective 7 N.E. Glucocorticoid 1 N.E. Glutamate, Non-Selective N.E. < 10 Glutamate, AMPA N.E. < 10 Glutamate, kainate N.E. < 10 Glutamate, NMDA, Agonist Site N.E. 10 Glutamate, NMDA, Phencyclidine Site N.E. < 10 Glutamate, Metabotropic, mGlu2 -1 N.E. Glutamate, Metabotropic, mGlu5 13 N.E. Glycine, Strychnine-Sensitive 5 N.E. Transporter, Glycine 0 N.E.
阻害率(%)は 1 回の実験(n=2)から算出した。 GABA: γ-aminobutyric acid AMPA: α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid NMDA: N-methyl-D-aspartic acid N.E.: 実施せず
- 45 -
ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 38
表 2.6.2-7. 各種受容体、イオンチャネルおよびトランスポーターへの特異的標識リガンド
結合に対する TRK-820 の阻害作用 (続き)
受容体/イオンチャネル/トランスポーター 阻害率 (%)
TRK-820 1000 nmol/L
TRK-820 10000 nmol/L
Histamine H1 N.E. < 10 Histamine H2 < 10 N.E. Histamine H3 < 10 N.E. Interleukin-1β (IL-1β) < 10 N.E. Interleukin-8 (IL-8) < 10 N.E. Leukotriene D4 (LTD4) < 10 N.E. Muscarinic M1 a 41 ± 3 72 ± 1 Muscarinic M2 N.E. < 10 Muscarinic M3 N.E. < 10 Neurokinin NK1 N.E. < 10 Neurokinin NK2 < 10 N.E. Neurokinin NK3 < 10 N.E. Neuropeptide Y2 N.E. < 10 Nicotinic Acetylcholine 5 N.E. Orphanin ORL1 47 N.E. Orexin OX1 25 N.E. Platelet activating factor (PAF) < 10 N.E. Potassium Channel [KA] 4 N.E. Potassium Channel [KATP] -14 N.E. Purinergic P2X -7 N.E. Purinergic P2Y 3 N.E. Ryanodine RyR3 2 N.E. Serotonin 5-HT1 N.E. < 10 Serotonin 5-HT1A N.E. < 10 Serotonin 5-HT2 N.E. 12 Serotonin 5-HT2A -6 N.E. Serotonin 5-HT3 N.E. < 10 Serotonin 5-HT4 24 N.E. Transporter, Serotonin (SERT) 4 N.E. Sigma, Non-Selective N.E. 14 ± 3 Somatostatin N.E. < 10 Tumor necrosis factor α (TNF-α) < 10 N.E. Vasoactive intestinal peptide (VIP) < 10 N.E. Vasopressin V1 < 10 < 10 Vasopressin V2 < 10 N.E.
阻害率(%)は 1 回の実験(n=2)から算出した(ただし Muscarnic M1受容体および Sigma(Non-Selective)受容体は 3 回の実
験(n=6)の平均値 ± 標準誤差を示す)。 N.E.: 実施せず a: Ki 値=1700 ± 200 nmol/L(3 回の実験の平均値 ± 標準誤差を示す)
- 46 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 39
(3) 引っ掻き行動抑制作用に対するオピオイドκ受容体拮抗薬の皮下投与の影響(in vivo)(2.6.3.2 (4)、4.2.1.1-22: 試験)
a) 方法
ICR 系雄性マウスの吻側背部皮内に、サブスタンス P(250 nmol/50 μL/site)を投与することに
よって誘発される引っ掻き行動を、サブスタンス P 投与直後から 30 分後まで測定した。サブスタ
ンス P 皮内投与の 30 分前に TRK-820(100 μg/kg)もしくは溶媒を経口投与し、引っ掻き行動の
抑制作用を指標に止痒作用を評価した。オピオイド κ 受容体拮抗薬の nor-BNI(1、3 および 10 mg/kg)
もしくは溶媒を、TRK-820 投与の 1 日前に皮下投与した。
b) 成績
nor-BNI の単独皮下投与は、いずれの用量でもサブスタンス P 誘発引っ掻き行動に影響を与えな
かった。一方、nor-BNI の前投与により TRK-820 の引っ掻き行動抑制作用は用量依存的に抑制さ
れた。nor-BNI の 10 mg/kg では、完全な拮抗作用を示さないものの、TRK-820 の引っ掻き行動に
対する統計学的に有意な抑制作用は消失した(図 2.6.2-9)。したがって、TRK-820 はオピオイド κ
受容体の活性化を介して止痒作用を発現する可能性が示唆された。
- 47 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 40
図 2.6.2-9. 引っ掻き行動抑制作用に対するオピオイド κ 受容体拮抗薬の皮下投与の影響
nor-BNI 皮下投与の翌日に引っ掻き行動を評価 TRK-820 経口投与から起痒剤投与までの時間: 30 分 起痒剤なし: PBS(溶媒)を 50 μL/site の容量で皮内投与 溶媒: 5 w/v%マンニトール水溶液(溶媒)を 10 mL/kg の容量で経口投与 nor-BNI 非投与群: 蒸留水(溶媒)を 10 mL/kg の容量で皮下投与 縦軸: 30 分間の引っ掻き行動回数(平均値 ± 標準誤差) 全群 n=8 ##p<0.01(Welch 検定) *p<0.05、**p<0.01(t 検定もしくは Welch 検定)
(4) 引っ掻き行動抑制作用に対するオピオイドκ受容体拮抗薬の脳室内投与の影響(in vivo)(2.6.3.2 (4)、4.2.1.1-23: 試験)
a) 方法
ddY 系雄性マウスの吻側背部皮内に、サブスタンス P(250 nmol/50 μL/site)を投与することに
よって誘発される引っ掻き行動を、サブスタンス P 投与直後から 30 分後まで測定した。サブスタ
ンス P 皮内投与の 30 分前に TRK-820(10 μg/kg)もしくは溶媒を皮下投与し、引っ掻き行動の抑
制作用を指標に止痒作用を評価した。オピオイド κ 受容体拮抗薬の nor-BNI(10 μg/site)もしくは
溶媒を、TRK-820 投与の 1 日前に脳室内投与した。
b) 成績
nor-BNI の単独脳室内投与は、サブスタンス P 誘発引っ掻き行動に影響を与えなかった。一方、
nor-BNI の脳室内投与により TRK-820 の引っ掻き行動抑制作用は完全に消失した(図 2.6.2-10)。
したがって、TRK-820 は中枢神経系のオピオイド κ 受容体の活性化を介して止痒作用を発現する
可能性が示唆された。
0 0 1 3 100
100
200
300
:起痒剤あり+ 溶媒
:起痒剤あり+ TRK-820(100 µg/kg, p.o.)
引っ掻き行動回数/30分
## ****
*
nor-BNI (mg/kg, s.c.)
:起痒剤なし+ 溶媒
起痒剤:サブスタンスP (250 nmol/site, i.d.)
- 48 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 41
図 2.6.2-10. 引っ掻き行動抑制作用に対するオピオイド κ 受容体拮抗薬の脳室内投与の
影響
nor-BNI 脳室内投与もしくは皮下投与の翌日に引っ掻き行動を評価 TRK-820 皮下投与から起痒剤投与までの時間: 30 分 起痒剤なし: PBS(溶媒)を 50 μL/site の容量で皮内投与 溶媒: 生理食塩液(溶媒)を 10 mL/kg の容量で皮下投与 nor-BNI 非投与群: 生理食塩液(溶媒)を 10 μL/site の容量で脳室内投与 縦軸: 30 分間の引っ掻き行動回数(平均値 ± 標準誤差) 全群 n=10 ##p<0.01(Welch 検定) **p<0.01(Dunnett 型多重比較)
(5) 局所麻酔作用の評価(in vivo)(2.6.3.2 (4)、4.2.1.1-24: 試験)
a) 方法
Hertley 系雄性モルモットの背部皮内に被験物質(容量 200 μL/site)を投与した。皮内投与の 5
分後から 30 分後まで、5 分ごとにマンドリン線(直径 0.2 mm)を用いて、皮内投与により生じた
丘疹の中央 1 ヵ所と周囲の 4 ヵ所の計 5 ヵ所を刺激した(5 ヵ所 × 6 回=30 回)。マンドリン線刺
激によって誘発される皮膚の攣縮反応を観察し、30 回の刺激による攣縮の総回数を記録し、攣縮
反応の抑制を指標に局所麻酔作用を評価した。TRK-820 は 0.01~10 μg/mL の濃度で皮内投与した。
局所麻酔薬プロカインは 2.5~10 mg/mL の濃度で皮内投与した。
- - + - + +0
25
50
75
100
:起痒剤あり+ 溶媒
:起痒剤あり+ TRK-820(10 µg/kg, s.c.)
引っ掻き行動回数/30分
##
****
起痒剤:サブスタンスP (250 nmol/site, i.d.)
nor-BNI(10 µg/site)
:起痒剤なし+ 溶媒
i.c.v. s.c.i.c.v.
nor-BNI (10 µg/animal)
- 49 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 42
b) 成績
TRK-820 のいずれの濃度を皮内投与しても、マンドリン線刺激による皮膚の攣縮反応は抑制さ
れなかった。一方、プロカインは、すべての濃度において用量依存的に攣縮反応を統計学的に有
意に抑制した(図 2.6.2-11)。したがって、TRK-820 は局所麻酔作用を有しておらず、止痒作用の
発現に局所麻酔作用が関与している可能性はないものと考えられた。
図 2.6.2-11. 局所麻酔作用の評価
TRK-820 とプロカインは 200 μL/site の容量で皮内投与 溶媒: 5 w/v%マンニトール水溶液を 200 μL/site の容量で皮内投与 縦軸: 5 ヵ所 × 6 回=30 回の刺激に対する攣縮反応の総回数(平均値 ± 標準誤差) 全群 n=4 **p<0.01 vs. 溶媒投与群(Dunnett 型多重比較)
(6) 経口剤(軟カプセル剤)中の不純物のヒトオピオイド受容体結合性(in vitro)(2.6.3.2 (5)、4.2.1.1-25: 試験)
経口剤中の不純物である 10α-OH のヒトオピオイド受容体に対する結合性を評価するために、
ヒトオピオイド受容体を発現させた培養細胞の細胞膜を用いた受容体結合試験を実施した。
a) 方法
ヒトオピオイド受容体を発現させた培養細胞(オピオイド κ 受容体:HEK-293 細胞、オピオイ
ド μ および δ 受容体:CHO 細胞)の膜標本を用いた。各受容体の特異的リガンドとして[3H]ジプ
レノルフィン(オピオイド κ および μ 受容体)または[3H]ナルトリンドール(オピオイド δ 受容
体)を使用し、10α-OH の各受容体の特異的リガンド結合に対する阻害作用を指標として、受容体
0 0.01 0.1 1 10 2.5 5 100
10
20
30
:プロカイン(200 µL/site, i.d.)
:TRK-820(200 µL/site, i.d.)
攣縮反応回数
**
**
溶液濃度(µg/mL)
:溶媒(200 µL/site, i.d.)
**
溶液濃度(mg/mL)
- 50 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 43
結合性を評価した。プライマリアッセイとして 10000 nmol/L の濃度での阻害作用を評価した。プ
ライマリアッセイで 50%以上の阻害作用を示した場合に、6 濃度を用いた実験を行い、Ki 値を算
出した。
b) 成績
プライマリアッセイの結果、10α-OH の 10000 nmol/L でのオピオイド κ、μ および δ 受容体に対
する特異的リガンド結合の阻害率は、それぞれ 100、66 および 8%であった(表 2.6.2-8)。そこで、
オピオイド κ および μ 受容体について 6 濃度を用いた実験を行い、Ki 値を算出した(表 2.6.2-8)。
10α-OH の濃度は 1~300 nmol/L(オピオイド κ 受容体)および 300~100000 nmol/L(オピオイド μ
受容体)とした。オピオイド κ 受容体に対する 10α-OH の Ki 値は 4.26 nmol/L であり、TRK-820
の17倍高値であった。オピオイドμ受容体に対する10α-OHのKi値は2070 nmol/Lであり、TRK-820
の 937 倍高値であった。
表 2.6.2-8. 経口剤中の不純物のオピオイド受容体結合性(Ki 値)
経口剤中の不純物 10α-OH 受容体タイプ オピオイド κ 受容体 オピオイド μ 受容体 オピオイド δ 受容体 結合阻害率 (%)a 100 66 8 Ki 値 (nmol/L)b 4.26 2070 > 10000 c
a: 10α-OH の 10000 nmol/L 適用時の阻害率。数値は 1 回の実験(n=2)から算出した。 b: 数値は 1 回の実験(n=2)から算出した。 c: 10α-OH の 10000 nmol/L 適用時の阻害率が 50%未満であったため、Ki 値を算出する実験は実施していない。
(7) 代謝物のヒトオピオイド受容体結合性(in vitro)(2.6.3.2 (6)、4.2.1.1-26: 試験)
代謝物である de-CPM、NFA-G および de-CPM-G のヒトオピオイド受容体に対する結合性を評
価するために、ヒトオピオイド受容体を発現させた培養細胞の細胞膜を用いた受容体結合試験を
実施した。
a) 方法
ヒトオピオイド受容体を発現させた培養細胞(オピオイド κ 受容体:HEK-293 細胞、オピオイ
ド μ 受容体:CHO-K1 細胞、オピオイド δ 受容体:CHO 細胞)の膜標本を用いた。各受容体の特
異的リガンドとして[3H]ジプレノルフィン(オピオイド κ および μ 受容体)または[3H]ナルトリン
ドール(オピオイド δ 受容体)を使用し、de-CPM、NFA-G および de-CPM-G の各受容体の特異的
リガンド結合に対する阻害作用を指標として、受容体選択性を評価した。プライマリアッセイと
して 10000 nmol/L の濃度での阻害作用を評価した。プライマリアッセイで 50%以上の阻害作用を
示した場合に、6 濃度を用いた実験を行い、Ki 値を算出した。
- 51 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 44
b) 成績
プライマリアッセイの結果、de-CPM の 10000 nmol/L でのオピオイド κ、μ および δ 受容体に対
する特異的リガンド結合の阻害率は、それぞれ103、93および25%であった。NFA-Gの10000 nmol/L
でのオピオイド κ、μ および δ 受容体に対する特異的リガンド結合の阻害率はそれぞれ 73、25 お
よび 3%であった。de-CPM-G の 10000 nmol/L でのオピオイド κ、μ および δ 受容体に対する特異
的リガンド結合の阻害率はそれぞれ 6、-1 および-8%であった(表 2.6.2-9)。
表 2.6.2-9. 代謝物のオピオイド受容体結合性(プライマリアッセイ)
代謝物名 (濃度 10000 nmol/L)
結合阻害率 (%)a オピオイド κ 受容体 オピオイド μ 受容体 オピオイド δ 受容体
de-CPM 103 93 25 NFA-G 73 25 3 de-CPM-G 6 -1 -8
a: 数値は 1 回の実験(n=2)から算出した。
プライマリアッセイで 50%以上の結合阻害作用を示した de-CPM(オピオイド κ および μ 受容
体)並びに NFA-G(オピオイド κ 受容体)について、Ki 値を算出するために 6 濃度を用いた実験
を行った。de-CPM の濃度は 0.3~100 nmol/L(オピオイド κ 受容体)および 30~10000 nmol/L(オ
ピオイド μ 受容体)とした。NFA-G の濃度は 100~30000 nmol/L(オピオイド κ 受容体)とした。
オピオイド κ 受容体に対する de-CPM の Ki 値は 5.95 nmol/L であり、TRK-820 の 24 倍高値であっ
た。オピオイド μ 受容体に対する de-CPM の Ki 値は 133 nmol/L であり、TRK-820 の 60 倍高値で
あった。また、オピオイド κ 受容体に対する NFA-G の Ki 値は 1960 nmol/L であり、TRK-820 の
8030 倍高値であった(表 2.6.2-10)。
表 2.6.2-10. 代謝物のオピオイド受容体結合性(Ki 値)
代謝物名 Ki 値 (nmol/L)a
オピオイド κ 受容体 オピオイド μ 受容体 オピオイド δ 受容体 de-CPM 5.95 ± 0.0643 133 ± 16.0 > 10000 b NFA-G 1960 ± 49.1 > 10000 b > 10000 b de-CPM-G > 10000 b > 10000 b > 10000 b
a: 数値は 3 回の実験の平均値 ± 標準誤差で示した。 b: 10000 nmol/L 適用時の阻害率が 50%未満であったため、Ki 値を算出する実験は実施していない。
- 52 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 45
(8) 経口剤(軟カプセル剤)中の不純物および代謝物のヒトオピオイド受容体作動性(in vitro)(2.6.3.2 (7)、4.2.1.1-27: 試験)
経口剤中の不純物である 10α-OH、代謝物である de-CPM、NFA-G および de-CPM-G のヒトオピ
オイド受容体作動性を、ヒトオピオイド受容体を発現させた CHO 細胞を用いたフォルスコリン刺
激による cAMP 産生に対する抑制作用を指標に評価した。
a) 方法
ヒトオピオイド受容体を発現させた培養細胞(オピオイド κ 受容体:CHO-K1 細胞、オピオイ
ドμおよび δ受容体:CHO-dhfr(-)細胞)を用いた。フォルスコリン 30 μmol/Lと共に 10α-OH、de-CPM、
NFA-G、de-CPM-G、TRK-820 または各受容体の標準的完全作動薬を各細胞に適用し、37°C で 30
分間インキュベーションした。産生された cAMP 量を AlphaScreenTM cAMP Assay kit および万能マ
イクロプレートアナライザー(Fusion αシステム)を用いて測定した。10α-OHは0.0003~100 nmol/L
(オピオイド κ 受容体)および 0.01~3000 nmol/L(オピオイド μ および δ 受容体)、de-CPM、NFA-G
および de-CPM-G は 0.01~3000 nmol/L(すべての受容体)、TRK-820 は 0.00003~10 nmol/L(オピ
オイド κ 受容体)および 0.01~3000 nmol/L(オピオイド μ および δ 受容体)、U-69593 は 0.001~
300 nmol/L(オピオイド κ 受容体)、DAMGO は 0.003~1000 nmol/L(オピオイド μ 受容体)、DPDPE
は 0.0003~100 nmol/L(オピオイド δ 受容体)の濃度範囲を用いた。
b) 成績
ヒトオピオイド κ受容体発現細胞においてフォルスコリン刺激誘発 cAMP産生に対する10α-OH、
de-CPM および NFA-G の最大抑制率(Imax)はそれぞれ 90、91 および 90%であり、TRK-820(Imax:
91%)と同等であったが、10α-OH、de-CPM および NFA-G の EC50はそれぞれ 0.0652、2.56 および
43.2 nmol/L であり、TRK-820(EC50:0.00940 nmol/L)のそれぞれ 6.9、272 および 4600 倍高濃度
であった。また、de-CPM-G は、最高濃度の 3000 nmol/L において最大抑制作用まで達しておらず、
抑制率も 22%であった(表 2.6.2-11)。したがって、経口剤中の不純物のオピオイド κ 受容体に対
する作用は TRK-820 よりも 6.9 倍弱く、代謝物のオピオイド κ 受容体に対する作用は TRK-820 と
比較して極めて弱いことが示唆された。
ヒトオピオイドμ受容体発現細胞ではNFA-Gおよびde-CPM-Gの Imaxはそれぞれ14および-5.5%
であり、TRK-820(Imax:50%)と比較し低値であり、ほとんど作用がなかった。また、10α-OH
および de-CPM は最高濃度の 3000 nmol/L において最大抑制作用まで達しておらず、抑制率はそれ
ぞれ 46 および 70%であり(表 2.6.2-11)、TRK-820 がほぼ最大抑制作用を発現している濃度
(30 nmol/L)においてはほとんど作用を示さなかった(図 2.6.2-12)。したがって、経口剤中の不
- 53 -
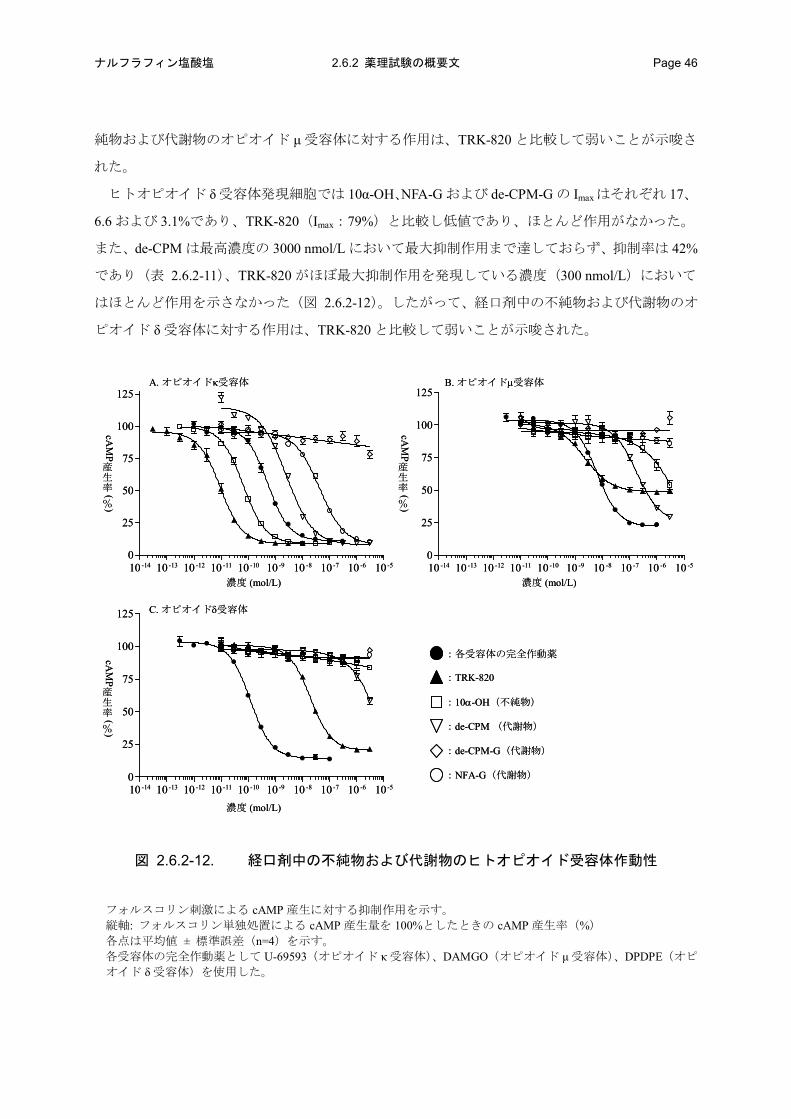
ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 46
純物および代謝物のオピオイド μ 受容体に対する作用は、TRK-820 と比較して弱いことが示唆さ
れた。
ヒトオピオイド δ 受容体発現細胞では 10α-OH、NFA-G および de-CPM-G の Imaxはそれぞれ 17、
6.6 および 3.1%であり、TRK-820(Imax:79%)と比較し低値であり、ほとんど作用がなかった。
また、de-CPM は最高濃度の 3000 nmol/L において最大抑制作用まで達しておらず、抑制率は 42%
であり(表 2.6.2-11)、TRK-820 がほぼ最大抑制作用を発現している濃度(300 nmol/L)において
はほとんど作用を示さなかった(図 2.6.2-12)。したがって、経口剤中の不純物および代謝物のオ
ピオイド δ 受容体に対する作用は、TRK-820 と比較して弱いことが示唆された。
図 2.6.2-12. 経口剤中の不純物および代謝物のヒトオピオイド受容体作動性
フォルスコリン刺激による cAMP 産生に対する抑制作用を示す。 縦軸: フォルスコリン単独処置による cAMP 産生量を 100%としたときの cAMP 産生率(%) 各点は平均値 ± 標準誤差(n=4)を示す。 各受容体の完全作動薬として U-69593(オピオイド κ 受容体)、DAMGO(オピオイド μ 受容体)、DPDPE(オピ
オイド δ 受容体)を使用した。
10 -14 10-13 10 -12 10 -11 10 -10 10-9 10 -8 10-7 10 -6 10-50
25
50
75
100
125
10 -14 10-13 10 -12 10 -11 10 -10 10-9 10 -8 10-7 10 -6 10-50
25
50
75
100
125
10 -14 10 -13 10 -12 10 -11 10-10 10-9 10 -8 10 -7 10 -6 10 -50
25
50
75
100
125
cA
MP
産生率(
%)
cAM
P
産生率(
%)
cAM
P産生率(
%)
A. オピオイドκ受容体 B. オピオイドµ受容体
C. オピオイドδ受容体
濃度 (mol/L) 濃度 (mol/L)
濃度 (mol/L)
:各受容体の完全作動薬
:TRK-820
:de-CPM (代謝物)
:de-CPM-G(代謝物)
:NFA-G(代謝物)
:10α-OH(不純物)
10 -14 10-13 10 -12 10 -11 10 -10 10-9 10 -8 10-7 10 -6 10-50
25
50
75
100
125
10 -14 10-13 10 -12 10 -11 10 -10 10-9 10 -8 10-7 10 -6 10-50
25
50
75
100
125
10 -14 10 -13 10 -12 10 -11 10-10 10-9 10 -8 10 -7 10 -6 10 -50
25
50
75
100
125
cA
MP
産生率(
%)
cAM
P
産生率(
%)
cAM
P産生率(
%)
A. オピオイドκ受容体 B. オピオイドµ受容体
C. オピオイドδ受容体
濃度 (mol/L) 濃度 (mol/L)
濃度 (mol/L)
:各受容体の完全作動薬
:TRK-820
:de-CPM (代謝物)
:de-CPM-G(代謝物)
:NFA-G(代謝物)
:10α-OH(不純物)
- 54 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 47
表 2.6.2-11. 経口剤中の不純物および代謝物のヒトオピオイド受容体作動性
被験物質 オピオイド κ 受容体 オピオイド μ 受容体 オピオイド δ 受容体 EC50
(nmol/L) Imax (%)
EC50 (nmol/L)
Imax (%)
EC50 (nmol/L)
Imax (%)
10α-OH (不純物) 0.0652 ± 0.0021 90.4 ± 0.7 N.C. (46.4 ± 3.4)b N.C.a 16.6 ± 1.2 de-CPM (代謝物) 2.56 ± 0.14 90.6 ± 0.5 N.C. (70.4 ± 1.5)b N.C. (41.6 ± 2.4)b NFA-G (代謝物) 43.2 ± 3.1 89.7 ± 0.6 N.C.a 14.1 ± 3.6 N.C.a 6.6 ± 1.5 de-CPM-G (代謝物) N.C. (21.7 ± 2.9)b N.C.a -5.5 ± 4.9 N.C.a 3.1 ± 1.1
TRK-820 0.00940 ± 0.00138 90.8 ± 0.6 2.35 ± 0.31 50.3 ± 1.8 19.7 ± 1.3 78.5 ± 1.4
各受容体の完全作動薬 c 0.525 ± 0.030 89.4 ± 0.7 6.49 ± 0.50 76.7 ± 1.1 0.119 ± 0.006 86.4 ± 0.3 数値は平均値 ± 標準誤差(n=4)を示す。 N.C.: 最高濃度において最大反応に達していなかったため、算出しなかった。 a: 最高濃度において最大反応に達していたが、作用が弱く、算出不能であった。 b: 最高濃度において最大反応に達していなかったため、参考値として最高濃度での反応率を示した。 c: U-69593(オピオイド κ 受容体)、DAMGO(オピオイド μ 受容体)、DPDPE(オピオイド δ 受容体)
(9) 代謝物のオピオイド受容体以外の受容体、イオンチャネルまたはトランスポーターに対す
る作用(in vitro)(2.6.3.2 (8)、4.2.1.1-28: 試験)
特異的リガンドの結合に対する代謝物である de-CPM、NFA-G および de-CPM-G の阻害作用を
指標として、各受容体、イオンチャネルまたはトランスポーターに対する結合性を評価した。
a) 方法
各受容体、イオンチャネルまたはトランスポーターへの結合試験に用いたリガンドと標本を示
した(表 2.6.2-12)。
- 55 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 48
表 2.6.2-12. 結合試験において用いた特異的標識リガンドおよび標本
受容体/イオンチャネル/ トランスポーター 標識リガンド 標本
Adenosine A1 [3H]DPCPX Human recombinant Adenosine A2A [3H]CGS-21680 Human recombinant Transporter, Adenosine [3H]Nitrobenzylthioinosine Guinea pig cerebral cortex Adrenergic α1, Non-Selective [3H]Prazosin Rat brain Adrenergic α2, Non-Selective [3H]Rauwolscine Rat cerebral cortex Adrenergic β1 [125I]Cyanopindolol Human recombinant Adrenergic β2 [3H]CGP-12177 Human recombinant Transporter, Norepinephrine (NET) [125I]RTI-55 Human recombinant Angiotensin AT2 [125I]CGP-42112A Human recombinant Atrial Natriuretic Factor (ANF) [125I]ANF (rat) Guinea pig adrenal gland Bombesin, Non-Selective [125I](Tyr4)-Bombesin Rat brain Bradykinin B2 [3H]Bradykinin Human recombinant Calcitonin Gene-Related Peptide CGRP1
[125I]CGRP (human) Human recombinant
Calcium Channel L-Type, Dihydropyridine [3H]Nitrendipine Rat cerebral cortex
Calcium Channel N-Type [125I]ω-Conotoxin GVIA Rat frontal brain Cannabinoid CB1 [3H]SR141716A Human recombinant Chemokine CCR1 [125I]MIP-1α Human recombinant Chemokine CCR2B [125I]MCP-1 Human recombinant Cholecystokinin CCK1 (CCKA) [3H]L-364,718 Human recombinant Cholecystokinin CCK2 (CCKB) [125I]CCK-8 Human recombinant Transporter, Choline [3H]Hemicholinium-3 Rat brain striatum Corticotropin Releasing Factor CRF1
[125I](Tyr0)-CRF (ovine) Human recombinant
Dopamine D1 [3H]SCH-23390 Human recombinant Dopamine D2L [3H]Spiperone Human recombinant Dopamine D3 [3H]Spiperone Human recombinant Transporter, Dopamine (DAT) [125I]RTI-55 Human recombinant Transporter, GABA [3H]GABA Rat cerebral cortex GABAA, Muscimol, Central [3H]Muscimol Rat brain (minus cerebellum) GABAA, Flunitrazepam, Central [3H]Flunitrazepam Rat brain (minus cerebellum) GABAA, Chloride Channel, TBPS [35S]TBPS Rat cerebral cortex GABAB, Non-Selective [3H]CGP-54626 Rat brain Glucocorticoid [3H]Dexamethasone Human recombinant Glutamate, AMPA [3H]AMPA Rat cerebral cortex Glutamate, Kainate [3H]Kainic acid Rat brain (minus cerebellum) Glutamate, NMDA, Agonism [3H]CGP-39653 Rat cerebral cortex Glutamate, NMDA, Phencyclidine [3H]TCP Rat cerebral cortex Glutamate, Non-Selective [3H]L-Glutamic acid Rat brain Glutamate, Metabotropic, mGlu2 [3H]LY341495 Human recombinant Glutamate, Metabotropic, mGlu5 [3H]Quisqualic acid Human recombinant
DPCPX: 8-cyclopentyl-1,3-dipropylxanthine MIP-1α: macrophage inflammatory protein-1α MCP-1: monocyte chemoattractant protein-1 GABA: γ-aminobutyric acid TBPS: t-butylbicyclophosphorothionate AMPA: α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid NMDA: N-methyl-D-aspartic acid TCP: tenocyclidine
- 56 -
ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 49
表 2.6.2-12. 結合試験において用いた特異的標識リガンドおよび標本 (続き)
受容体/イオンチャネル/ トランスポーター 標識リガンド 標本
Glycine, Strychnine-Sensitive [3H]Strychnine Rat spinal cord Transporter, Glycine [3H]Glycine Rat brain cortex Histamine H1 [3H]Pyrilamine Human recombinant Histamine H2 [125I]Aminopotentidine Human recombinant
Histamine H3 [3H]N-α-Methylhistamine (NAMH) Human recombinant
Interleukin IL-1 [125I]Interleukin-1β Mouse 3T3-SWISS cells Chemokine CXCR1/2 (IL-8, Non-Selective) [125I]IL-8 Human neutrophils
Leukotriene, Cysteinyl CysLT1 [3H]LTD4 Human recombinant Muscarinic M1 [3H]N-Methylscopolamine Human recombinant Muscarinic M2 [3H]N-Methylscopolamine Human recombinant Muscarinic M3 [3H]N-Methylscopolamine Human recombinant Tachykinin NK1 [3H]Substance P Human recombinant Tachykinin NK2 [3H]SR-48968 Human recombinant Tachykinin NK3 [125I]MePhe7-Neurokinin B Human recombinant Neuropeptide Y Y2 [125I]Peptide YY Human KAN-TS cells Nicotinic Acetylcholine [125I]Epibatidine Human IMR-32 cells Orphanin ORL1 [3H]Nociceptin Human recombinant Orexin OX1 [125I]Orexin A Human recombinant Platelet Activating Factor (PAF) [3H]PAF Human platelets Potassium Channel [KA] [125I]α-Dendrotoxin Rat cerebral cortex Potassium Channel [KATP] [3H]Glyburide Hamster pancreatic HIT-T15 beta cells Purinergic P2X [3H]α, β-Methylene ATP Rabbit urinary bladder Purinergic P2Y [35S]ATP-αS Rat brain Ryanodine RyR3 [3H]Ryanodine Rat cerebral cortex Serotonin 5-HT1, Non-Selective [3H]Serotonin (5-HT) Rat cerebral cortex Serotonin 5-HT1A [3H]8-OH-DPAT Human recombinant Serotonin 5-HT2, Non-Selective [3H]Ketanserin Rat brain Serotonin 5-HT2A [3H]Ketanserin Human recombinant Serotonin 5-HT3 [3H]GR-65630 Human recombinant Serotonin 5-HT4 [3H]GR-113808 Guinea pig striatum Transporter, Serotonin (SERT) [3H]Paroxetine Human recombinant Sigma, Non-Selective [3H]DTG Guinea pig brain Somatostatin sst1 [125I]Tyr11-Somatostatin 14 Human recombinant Tumor Necrosis Factor (TNF), Non-Selective [125I]TNF-α Human U937 cells
Vasoactive Intestinal Peptide VIP1 [125I]VIP Human HT29 colon adenocarcinoma cells
Vasopressin V1A [125I]PhenylacetylTyr(Me)PheGln AsnArgProArgTyr Human recombinant
Vasopressin V2 [3H](Arg8)-Vasopressin Human recombinant LTD4: leukotriene D4
ATP: adenosine triphosphate 8-OH-DPAT: 8-hydroxy-2(di-n-propylamino)-tetraline DTG: 1,3-di-(2-tolyl)-guanidine
- 57 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 50
b) 成績
de-CPM は L 型 Ca2+チャネルに対する[3H]ニトレンジピンの結合を 18%、コリントランスポータ
ーに対する[3H]ヘミコリニウム-3 の結合を 17%、GABAA受容体の GABA 結合部位、ベンゾジア
ゼピン結合部位および Cl-チャネルに対する[3H]ムシモール、[3H]フルニトラゼパムおよび
[35S]TBPSの結合をそれぞれ 11、13および 16%、グルタミン酸mGluR2受容体に対する[3H]LY341495
の結合を 12%、グリシン受容体に対する[3H]ストリキニーネの結合を 11%、ケモカイン CXCR1/2
受容体に対する[125I]IL-8 の結合を 12%、ムスカリン M1および M3受容体に対する[3H]N-メチルス
コポラミンの結合をそれぞれ 11 および 12%、プリン P2X受容体に対する[3H]α, β-メチレン ATP の
結合 12%を、プリン P2Y受容体に対する[35S]ATP-αS の結合を 18%およびセロトニン 5-HT4 受容体
に対する[3H]GR-113808 の結合を 23%阻害した。
また、NFA-G はアドレナリン α2 受容体に対する[3H]ラウオルシンの結合を 14%、ボンベシン受
容体に対する[125I](Tyr4)-ボンベシンの結合を 17%、ブラジキニン B2受容体に対する[3H]ブラジキ
ニンの結合を 10%、L 型 Ca2+チャネルに対する[3H]ニトレンジピンの結合を 13%、グリシントラ
ンスポーターに対する[3H]グリシンの結合を 18%、ヒスタミン H2 受容体に対する[125I]アミノポテ
ンチジンの結合を 10%、ケモカイン CXCR1/2 受容体に対する[125I]IL-8 の結合を 17%、ムスカリン
M3 受容体に対する[3H]N-メチルスコポラミンの結合を 10%およびプリン P2Y 受容体に対する
[35S]ATP-αS の結合を 22%阻害した。
さらに、de-CPM-G はアドレナリン α2受容体に対する[3H]ラウオルシンの結合を 12%、ブラジ
キニン B2受容体に対する[3H]ブラジキニンの結合を 17%、L 型 Ca2+チャネルに対する[3H]ニトレ
ンジピンの結合を 23%、コレシストキニン CCK1 受容体に対する[3H]L-364,718 の結合を 11%、ド
パミン D3 受容体に対する[3H]スピペロンの結合を 15%、GABA トランスポーターに対する
[3H]GABA の結合を 14%、GABAA受容体ベンゾジアゼピン結合部位に対する[3H]フルニトラゼパ
ムの結合を 20%、GABAB受容体に対する[3H]CGP-54626 の結合を 11%、グルタミン酸 AMPA およ
び mGluR2受容体に対する[3H]AMPA および[3H]LY341495 の結合をそれぞれ 11 および 10%、グリ
シントランスポーターに対する[3H]グリシンの結合を 10%、ヒスタミン H2受容体に対する[125I]ア
ミノポテンチジンの結合を 11%、ケモカイン CXCR1/2 受容体に対する[125I]IL-8 の結合を 23%、タ
キキニン NK1 受容体に対する[3H]サブスタンス P の結合を 11%、プリン P2Y 受容体に対する
[35S]ATP-αS の結合を 31%、セロトニン 5-HT2 および 5-HT4受容体に対する[3H]ケタンセリンおよ
び[3H]GR-113808 の結合をそれぞれ 13 および 15%阻害した。したがって、TRK-820 の代謝物は上
記の受容体、イオンチャネルもしくはトランスポーターに対して低い結合性を示したが、TRK-820
のオピオイド κ 受容体に対する結合性(ヒトオピオイド受容体結合試験における Ki 値:
0.244 nmol/L、表 2.6.2-3)と比較すると著しく低かった。その他の各受容体、イオンチャネルお
- 58 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 51
よびトランスポーターに対する各特異的リガンド結合に対する TRK-820 の代謝物の 1000 nmol/L
での阻害率はいずれも 10%未満であり、結合性はほとんど認められなかった(表 2.6.2-13)。
表 2.6.2-13. 各種受容体、イオンチャネルおよびトランスポーターへの特異的標識リガンド
結合に対する TRK-820 の代謝物の阻害作用
受容体/イオンチャネル/トランスポーター 阻害率 (%)
de-CPM 1000 nmol/L
NFA-G 1000 nmol/L
de-CPM-G 1000 nmol/L
Adenosine A1 2 -8 0 Adenosine A2A 7 0 3 Transporter, Adenosine -1 1 3 Adrenergic α1, Non-Selective 0 -4 2 Adrenergic α2, Non-Selective 9 14 12 Adrenergic β1 -2 1 4 Adrenergic β2 9 4 5 Transporter, Norepinephrine (NET) 6 6 3 Angiotensin AT2 -2 -2 -2 Atrial Natriuretic Factor (ANF) -2 2 -4 Bombesin, Non-Selective 8 17 8 Bradykinin B2 4 10 17 Calcitonin Gene-Related Peptide CGRP1 2 -2 4 Calcium Channel L-Type,Dihydropyridine 18 13 23 Calcium Channel N-Type 2 -5 2 Cannabinoid CB1 2 -14 2 Chemokine CCR1 2 -2 -1 Chemokine CCR2B -6 -4 -6 Cholecystokinin CCK1 (CCKA) 6 -5 11 Cholecystokinin CCK2 (CCKB) -5 -2 1 Transporter, Choline 17 9 1 Corticotropin Releasing Factor CRF1 -11 -5 1 Dopamine D1 -1 -9 -3 Dopamine D2L 3 -3 1 Dopamine D3 5 1 15 Transporter, Dopamine (DAT) -3 1 3 Transporter, GABA 2 -6 14 GABAA, Muscimol, Central 11 4 8 GABAA, Flunitrazepam, Central 13 0 20 GABAA, Chloride Channel, TBPS 16 7 9 GABAB, Non-Selective 4 7 11 Glucocorticoid 3 9 9
阻害率(%)は 1 回の実験(n=2)から算出した。
- 59 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 52
表 2.6.2-13. 各種受容体、イオンチャネルおよびトランスポーターへの特異的標識リガンド
結合に対する TRK-820 の代謝物の阻害作用 (続き)
受容体/イオンチャネル/トランスポーター 阻害率 (%)
de-CPM 1000 nmol/L
NFA-G 1000 nmol/L
de-CPM-G 1000 nmol/L
Glutamate, AMPA -8 -6 11 Glutamate, Kainate -7 -11 1 Glutamate, NMDA, Agonism 6 5 4 Glutamate, NMDA, Phencyclidine -4 -8 -4 Glutamate, Non-Selective 0 1 7 Glutamate, Metabotropic, mGlu2 12 -5 10 Glutamate, Metabotropic, mGlu5 -16 -7 0 Glycine, Strychnine-Sensitive 11 -5 -17 Transporter, Glycine 3 18 10 Histamine H1 -6 -5 -9 Histamine H2 7 10 11 Histamine H3 0 5 4 Interleukin IL-1 7 1 2 Chemokine CXCR1/2 (IL-8, Non-Selective) 12 17 23 Leukotriene, Cysteinyl CysLT1 -4 -1 0 Muscarinic M1 11 -10 -18 Muscarinic M2 8 -8 6 Muscarinic M3 12 10 6 Tachykinin NK1 1 -6 11 Tachykinin NK2 -3 -12 -3 Tachykinin NK3 -9 -3 -5 Neuropeptide Y Y2 -4 1 3 Nicotinic Acetylcholine 9 2 -5 Orphanin ORL1 3 -5 5 Orexin OX1 4 4 6 Platelet Activating Factor (PAF) -1 -7 -6 Potassium Channel [KA] 0 7 -1 Potassium Channel [KATP] 2 -3 3 Purinergic P2X 12 0 -4 Purinergic P2Y 18 22 31 Ryanodine RyR3 0 2 0 Serotonin 5-HT1, Non-Selective 6 -1 1 Serotonin 5-HT1A -3 -7 6 Serotonin 5-HT2, Non-Selective 0 1 13 Serotonin 5-HT2A 1 -2 -1 Serotonin 5-HT3 -5 0 4 Serotonin 5-HT4 23 -9 15 Transporter, Serotonin (SERT) 5 -9 -3 Sigma, Non-Selective -8 -5 -4 Somatostatin sst1 3 1 -4 Tumor Necrosis Factor (TNF), Non-Selective 5 9 9 Vasoactive Intestinal Peptide VIP1 -2 4 -3 Vasopressin V1A 4 5 -4 Vasopressin V2 3 -4 4
阻害率(%)は 1 回の実験(n=2)から算出した。
- 60 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 53
(10) 代謝物のサブスタンスP誘発引っ掻き行動に対する作用(in vivo)(2.6.3.2 (9)、4.2.1.1-29:試験、4.2.1.1-30: 試験、4.2.1.1-31: 試験、4.2.1.1-32:試験)
代謝物である de-CPM、NFA-G および de-CPM-G の止痒作用を評価した。
a) 方法
ddY 系雄性マウスの吻側背部皮内に、サブスタンス P(250 nmol/50 μL/site)を投与することに
よって誘発される引っ掻き行動を、サブスタンス P 投与直後から 30 分後まで測定した。サブスタ
ンス P 皮内投与の 30 分前に TRK-820(0.3~10 μg/kg)、de-CPM·T(1~1000 μg/kg)、NFA-G(1~
1000 μg/kg)または de-CPM-G(1~1000 μg/kg)を皮下投与し、引っ掻き行動の抑制作用を指標に
止痒作用を評価した。なお、de-CPM は、溶媒として使用した 5 w/v%マンニトール水溶液(本評
価に含まれる 4 試験共通の溶媒)への溶解性が低いため、de-CPM の酒石酸塩である de-CPM·T を
使用した。
b) 成績
TRK-820 は用量依存的に引っ掻き行動を抑制し、10 μg/kg において統計学的に有意な作用を示
した(図 2.6.2-13)。ED50は、1.65 μg/kg(95%信頼区間:0.880~3.08 μg/kg)であった。de-CPM·T、
NFA-G または de-CPM-G を、未変化体である TRK-820 が統計学的に有意な引っ掻き行動抑制作用
を示した 10 μg/kg の 100 倍量である 1000 μg/kg まで皮下投与しても、統計学的に有意な引っ掻き
行動抑制作用は観察されなかった(図 2.6.2-13)。以上のように、全身性に投与された TRK-820
の代謝物 de-CPM、NFA-G および de-CPM-G は止痒作用を発現しない可能性が示唆されたことか
ら、経口投与による TRK-820 の止痒作用発現に代謝物は寄与していないことが考えられた。
- 61 -
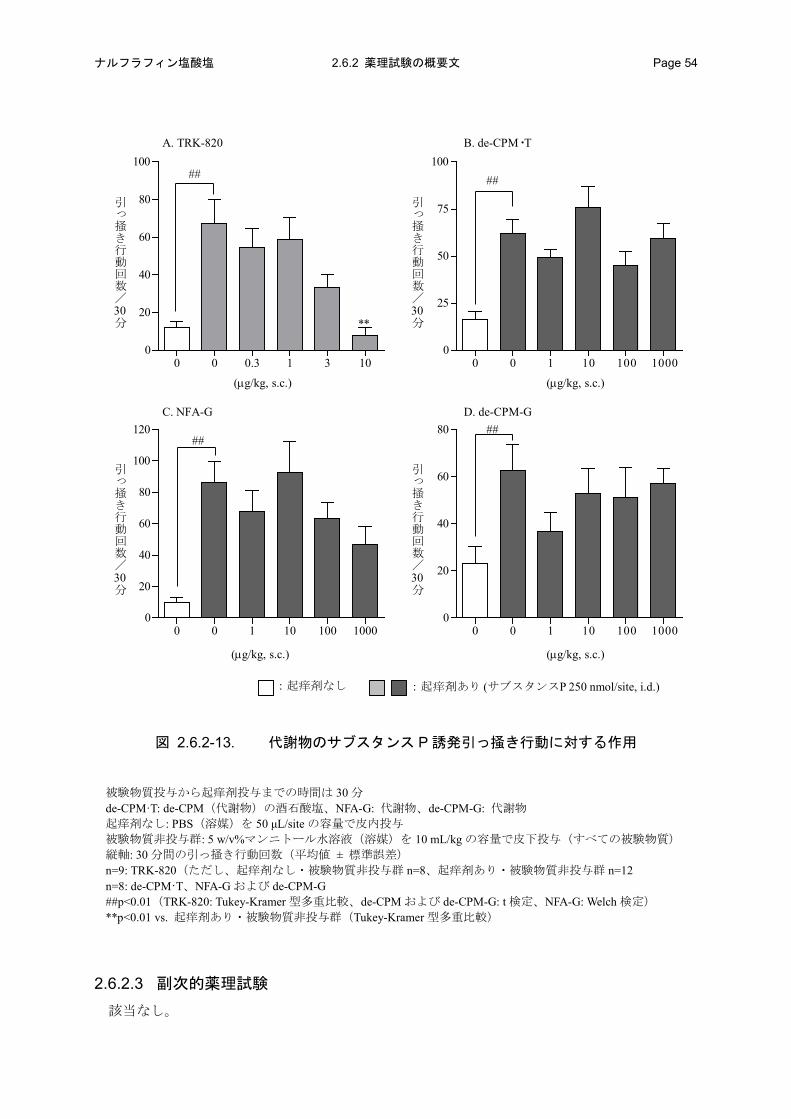
ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 54
図 2.6.2-13. 代謝物のサブスタンス P 誘発引っ掻き行動に対する作用
被験物質投与から起痒剤投与までの時間は 30 分 de-CPM·T: de-CPM(代謝物)の酒石酸塩、NFA-G: 代謝物、de-CPM-G: 代謝物 起痒剤なし: PBS(溶媒)を 50 μL/site の容量で皮内投与 被験物質非投与群: 5 w/v%マンニトール水溶液(溶媒)を 10 mL/kg の容量で皮下投与(すべての被験物質) 縦軸: 30 分間の引っ掻き行動回数(平均値 ± 標準誤差) n=9: TRK-820(ただし、起痒剤なし・被験物質非投与群 n=8、起痒剤あり・被験物質非投与群 n=12 n=8: de-CPM·T、NFA-G および de-CPM-G ##p<0.01(TRK-820: Tukey-Kramer 型多重比較、de-CPM および de-CPM-G: t 検定、NFA-G: Welch 検定) **p<0.01 vs. 起痒剤あり・被験物質非投与群(Tukey-Kramer 型多重比較)
2.6.2.3 副次的薬理試験
該当なし。
0 0 1 10 100 10000
20
40
60
80
0 0 1 10 100 10000
20
40
60
80
100
120
0 0 1 10 100 10000
25
50
75
100
0 0 0.3 1 3 100
20
40
60
80
100A. TRK-820 B. de-CPM T
C. NFA-G
:起痒剤なし :起痒剤あり (サブスタンスP 250 nmol/site, i.d.)
引っ掻き行動回数/30分
引っ掻き行動回数/30分
引っ掻き行動回数/30分
(µg/kg, s.c.) (µg/kg, s.c.)
(µg/kg, s.c.)
##
**
##
##
引っ掻き行動回数/30分
##D. de-CPM-G
(µg/kg, s.c.)
・
- 62 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 55
2.6.2.4 安全性薬理試験
TRK-820 は、国内でも当初注射剤を用いて筋肉内投与で術後疼痛に対する開発が進められてい
たこともあり、安全性薬理試験については、「安全性薬理試験ガイドラインについて」(平成 13 年
6 月 21 日 医薬審発第 902 号)発出前であったため、「新医薬品等の製造(輸入)承認申請に必
要な一般薬理試験のガイドラインについて」(平成 3 年 1 月 29 日 薬新薬第 4 号)に準じて、い
くつかの試験で経口投与以外の投与経路(筋肉内、皮下、静脈内)による一般薬理試験を実施し
ていた(GLP 非準拠)。その後、そう痒症の治療を目的に、経口剤を開発することになったため、
経口投与で実施されていなかった試験を含む以下の試験について「新医薬品等の製造(輸入)承
認申請に必要な一般薬理試験のガイドラインについて」(平成 3 年 1 月 29 日 薬新薬第 4 号)も
しくは「安全性薬理試験ガイドラインについて」(平成 13 年 6 月 21 日 医薬審発第 902 号)に基
づき GLP 準拠で追加実施した。
1) hERG 電流阻害作用
2) モルモット摘出乳頭筋の活動電位に対する作用
3) 覚醒イヌを用いた心血管系および呼吸系への影響
4) 麻酔イヌを用いた心血管系への影響
5) マウスに対する麻酔作用(一般薬理試験のガイドライン)
6) ラット脳波に対する作用
7) マウスに対する痙攣誘発および抗痙攣作用(一般薬理試験のガイドライン)
8) 腎/泌尿器系に及ぼす影響(一般薬理試験のガイドライン)
9) 自律神経系に及ぼす影響
なお、各試験は上記の「安全性薬理試験ガイドラインについて」に基づき、安全性薬理コアバ
ッテリー試験、安全性薬理コアバッテリーに対するフォローアップ試験および補足的安全性薬理
試験に分類し、記載した。
さらに、TRK-820 は中枢神経系への副作用が懸念されたため、「ICR 系マウスにおける TRK-820
の自発運動抑制作用の評価 [TRK-820]」(2.6.3.4 (2) 1) b)、4.2.1.3-5: 試験)を自発運動
に対する作用(in vivo)に記載した(2.6.2.4 (2) 1) b))。なお、本試験は、「安全性薬理試験ガイド
ラインについて」(平成 13 年 6 月 21 日 医薬審発第 902 号)の発出後に実施したが、当該試験の
主目的が、効力を裏付ける試験として実施したマウスの引っ掻き行動抑制作用が、自発運動の低
下によるものではないことを確認するためであったため、GLP 非準拠として実施した。また、サ
ルの一般症状、行動および自発運動に対する作用について、安全性薬理コアバッテリーに対する
フォローアップ試験として記載した。なお、本試験はサルを用いた静脈内自己投与法による精神
依存性評価の予備試験として、GLP 準拠で実施した試験である。
- 63 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 56
(1) 安全性薬理コアバッテリー試験
1) 中枢神経系に及ぼす影響
a) ラットの一般症状および行動に及ぼす影響(in vivo)(2.6.3.4 (1) 1) a)、4.2.1.3-1:試験、GLP非準拠)
Wistar 系雄性ラットに TRK-820(100、300、1000 および 3000 μg/kg)を経口投与し、0.5、1、2、
4、6 および 8 時間後に Irwin の多次元観察法に準じて評価した。
TRK-820 は、100 および 300 μg/kg の経口投与ではラットの一般症状に対して何ら影響を及ぼさ
なかった。1000 μg/kg では身づくろいの軽度な低下、自発運動の軽度な低下、体温の軽度な低下
および軽度な眼瞼下垂が認められた。3000 μg/kg では警戒性の軽度な低下、身づくろいの軽度な
低下、反応性の軽度な低下、自発運動の軽度な低下、軽度なよろめき歩行、軽度な眼瞼下垂、体
温の軽度な低下ないし低下、軽度な四肢伸長、軽度な流涙、逃避反応の軽度な低下、痛覚反応の
軽度な低下、耳介反射の軽度な低下、軽度な腹這い姿勢、軽度な異常歩行および軽度な縮瞳が観
察された。
2) 心血管系に及ぼす影響
a) hERG電流に対する作用(in vitro)(2.6.3.4 (1) 2) a)、4.2.1.3-2: 試験、GLP準拠)
hERG 遺伝子を導入した HEK-293 細胞を用いて TRK-820(0、3、30、300 および 3000 nmol/L)
を適用してパッチクランプ法により hERG 電流を測定した。hERG 電流に対する無作用量は
3 nmol/L で、IC50 は 840 nmol/L であった。
b) 覚醒イヌを用いた試験(in vivo)(2.6.3.4 (1) 2) b)、4.2.1.3-3: 試験、GLP準拠)
イヌに TRK-820(0、3、10、100 および 300 μg/kg)を経口投与して無麻酔、非拘束で血圧(収
縮期血圧、拡張期血圧および平均血圧)、心拍数および心電図(PR 間隔、QRS 時間、QT 間隔お
よび QTc)を投与前、投与後 1、2、4、6 および 24 時間に測定した。血圧では、収縮期血圧およ
び平均血圧共に TRK-820 の 10 μg/kg 投与で低下し、100 および 300 μg/kg 投与では軽度な血圧低
下が認められた。心拍数は 10 μg/kg 以上の投与で増加した。心電図では PR 間隔、QRS 時間、QT
間隔および QTc 共にいずれの投与量でも影響は認められなかった。以上のことから、イヌにおけ
る心血管系に対する無作用量は 3 μg/kg と考えられた。
3) 呼吸系に及ぼす影響
a) 覚醒イヌを用いた試験(in vivo)(2.6.3.4 (1) 3) a)、4.2.1.3-3: 試験、GLP準拠)
イヌに TRK-820(0、3、10、100 および 300 μg/kg)を経口投与して無麻酔、非拘束で呼吸数、
動脈血中の pO2、pCO2、pH およびヘモグロビン O2 saturation を投与前、投与後 1、2、4、6 およ
- 64 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 57
び 24 時間に測定した。TRK-820 は 300 μg/kg でも全評価項目に影響を及ぼさなかった。以上のこ
とから、イヌにおける呼吸系に対する無作用量は 300 μg/kg より大きいと考えられた。
(2) 安全性薬理コアバッテリーに対するフォローアップ試験
1) 中枢神経系に及ぼす影響
a) サルの一般症状および行動に及ぼす影響(in vivo)(2.6.3.4 (2) 1) a)、4.2.3.7.4-5:試験、GLP準拠)
雌雄アカゲザルに TRK-820(0.25、0.5、1 および 2 μg/kg)を静脈内投与し、投与前、投与後 0.25、
0.5、1、2、3、4 および 5 時間に一般症状および行動を観察した。なお、投与 5 時間後の観察で投
与前と比較して変化が認められた場合、24 時間後の観察を実施した。
溶媒である 5 w/v%マンニトール水溶液を投与したところ、6 例中 5 例で観察者への攻撃行動の
増強もしくは減弱が認められ、そのうち 1 例では観察者への怯え表情の減弱が認められた。これ
らの多くは一過性の変化であった。TRK-820 の 0.25 μg/kg 静脈内投与では 2 例中 2 例共に変化は
認められなかった。0.5 μg/kg 静脈内投与では 2 例中 2 例で運動低下、うずくまり姿勢および観察
者への攻撃行動の増強、同 2 例中 1 例で観察者への攻撃行動の減弱、腹臥位および動作緩慢が観
察された。1 μg/kg 静脈内投与では 2 例中 2 例で観察者への攻撃行動の減弱、同 2 例中 1 例で運動
低下、うずくまり姿勢、腹臥位、観察者への怯え表情の減弱、動作緩慢および閉眼が観察された。
2 μg/kg 静脈内投与では 2 例中 2 例で流涎、運動低下、うずくまり姿勢、閉眼、動作緩慢および口
の半開状態、同 2 例中 1 例で観察者への攻撃行動の減弱、腹臥位および運動失調が認められた。
b) 自発運動に対する作用(in vivo)(2.6.3.4 (2) 1) b)、4.2.1.3-5: 試験、4.2.1.3-1:試験、4.2.1.3-6: 試験、4.2.1.3-7: 試験、GLP非準拠)
ICR 系雄性マウスに TRK-820(50、100、200 および 400 μg/kg)を経口投与し、30 分後から 30
分の移所運動を測定したところ、400 μg/kg で統計学的に有意に運動量が減少し(ED50:345 μg/kg、
95%信頼区間:258~627 μg/kg)、自発運動に対する抑制作用が観察された(2.6.3.4 (2) 1) b)、
4.2.1.3-5: 試験、GLP 非準拠)。Wistar 系雄性ラットに TRK-820(100、300、1000 およ
び 3000 μg/kg)を経口投与し、直後から 4 時間後まで、30 分ごとの移所運動を測定したところ、
1000 および 3000 μg/kg で統計学的に有意な自発運動抑制作用が観察された(2.6.3.4 (2) 1) b)、
4.2.1.3-1: 、GLP 非準拠)。
回転かご試験において、ICR 系雄性マウスに TRK-820(10、30、100 および 300 μg/kg)を経口
投与し、運動量を 30 分後から 30 もしくは 60 分測定したところ、いずれも 100 および 300 μg/kg
で統計学的に有意な自発運動の抑制作用が認められた(30 分測定時の ED50:103 μg/kg、95%信頼
区間:64.9~163 μg/kg、60 分測定時の ED50:90.6 μg/kg、95%信頼区間:54.5~151 μg/kg)
(2.6.3.4 (2) 1) b)、4.2.1.3-6: 試験、GLP 非準拠)。同じく回転かご試験において、ddY
- 65 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 58
系雄性マウスに TRK-820(1、3、10 および 30 μg/kg)を皮下投与し、運動量を 30 分後から 60 分
測定したところ、10 および 30 μg/kg で統計学的に有意な自発運動の抑制作用が認められた(ED50:
7.79 μg/kg、95%信頼区間:3.82~15.9 μg/kg)(2.6.3.4 (2) 1) b)、4.2.1.3-7: 試験、GLP 非
準拠)。
c) 協調運動に対する作用(in vivo)(2.6.3.4 (2) 1) c)、4.2.1.3-8: 試験、GLP非準拠)
ddY 系雄性マウスに TRK-820(10、30、100 および 300 μg/kg)を経口投与し、ロタロッド試験
により評価した。投与 30 分後では、300 μg/kg で統計学的に有意な影響が認められた(ED50:
115 μg/kg、95%信頼区間:68.6~194 μg/kg)。投与 60 分後では、100 および 300 μg/kg で統計学的
に有意な影響が認められた(ED50:72.3 μg/kg、95%信頼区間:44.0~119 μg/kg)。
d) 麻酔作用(in vivo)(2.6.3.4 (2) 1) d)、4.2.1.3-9: 試験、GLP準拠)
ICR 系雄性マウスに TRK-820(30、100、300、1000 および 3000 μg/kg)を経口投与し、30 分後
にペントバルビタール(50 mg/kg)を腹腔内投与し、ペントバルビタール誘発睡眠時間に対する
影響を評価した。1000 および 3000 μg/kg では、睡眠時間は溶媒対照群のそれぞれ 182 および 205%
となり、統計学的に有意な睡眠時間の延長が観察された。
e) 脳波に対する作用(in vivo)(2.6.3.4 (2) 1) e)、4.2.1.3-10: 試験、GLP準拠)
Wistar 系雄性ラットに TRK-820(3、10 および 30 μg/kg)を皮下投与し、無麻酔・非拘束下で自
発脳波を投与 1 時間前から 5 時間後まで測定した。脳波の波形に対しては、いずれの用量におい
ても明らかな変化は認められなかった。新皮質前頭葉の脳波の周波数解析に対しては、3 μg/kg で
アルファ帯域の減少、10 および 30 μg/kg でアルファおよびベータ-1 帯域の減少が認められた。海
馬の周波数解析に対しては、10 μg/kg でアルファ帯域の減少およびベータ-2 帯域の増加、30 μg/kg
でアルファ帯域の減少が認められた。睡眠-覚醒周期の各脳波水準に対しては、3 μg/kg で覚醒期の
増加、徐波睡眠期の減少および速波睡眠期の減少、10 μg/kg で覚醒期の増加、徐波睡眠期の減少、
速波睡眠期の減少および速波睡眠期の潜時時間の延長、30 μg/kg で覚醒期の増加、徐波睡眠期の
減少、速波睡眠期の減少、徐波睡眠期の潜時時間の延長および速波睡眠期の潜時時間の延長が統
計学的に有意な変化として認められた。
f) 鎮痛作用(in vivo)(2.6.3.4 (2) 1) f)、4.2.1.3-11: 試験、GLP非準拠)
ddY 系雄性マウスに TRK-820(12.5、25、50 および 100 μg/kg)を経口投与し、30 分後に酢酸ラ
イジング法により評価した。25 μg/kg以上で統計学的に有意なライジング回数の減少が観察され、
鎮痛作用を発現することが示唆された(ED50:31.9 μg/kg、95%信頼区間:25.1~40.6 μg/kg)。な
- 66 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 59
お、TRK-820(1.25、2.5、5 および 10 μg/kg)を皮下投与し、30 分後に測定した場合には、2.5 μg/kg
以上で統計学的に有意なライジング回数の減少が観察され、鎮痛作用を発現することが示唆され
た(ED50:3.29 μg/kg、95%信頼区間:2.52~4.29 μg/kg)。
g) 痙攣誘発および抗痙攣作用(in vivo)(2.6.3.4 (2) 1) g)、4.2.1.3-12: 試験、GLP準拠)
ICR 系雄性マウスに TRK-820(100、300、1000 および 3000 μg/kg)を経口投与し、30 分後に電
撃もしくはペンチレンテトラゾールを与えた。TRK-820 は、閾値下最大電撃および痙攣誘発閾値
より低用量のペンチレンテトラゾール皮下投与による強直性痙攣の発現数、間代性痙攣の発現数
および死亡数に影響を及ぼさなかった。また、TRK-820 は、最大電撃および痙攣誘発閾値以上の
用量のペンチレンテトラゾール皮下投与による強直性痙攣の発現数、間代性痙攣の発現数および
死亡数に影響を及ぼさなかった。
h) 正常体温に対する作用(in vivo)(2.6.3.4 (2) 1) h)、4.2.1.3-1: 試験、GLP非準拠)
Wistar 系雄性ラットに TRK-820(100、300、1000 および 3000 μg/kg)を経口投与し、直腸温を
投与前、投与 0.5、1、2、4、6、8、24 および 48 時間後に測定した。100 および 300 μg/kg では、
体温にはほとんど影響が認められなかった。1000 μg/kg では溶媒対照群と比較して 0.3~0.5°C の
軽度な体温低下が認められた。3000 μg/kg では 1.2~1.5°C の体温低下が認められた。
2) 心血管系に及ぼす影響
a) 摘出乳頭筋の活動電位に対する作用(in vitro)(2.6.3.4 (2) 2) a)、4.2.1.3-13: 試
験、GLP準拠)
Hartley 系雄性モルモットの心臓から摘出した乳頭筋を用いて TRK-820 を 0、30、300 および
3000 nmol/L で適用して活動電位を測定した。静止膜電位、活動電位振幅、APD20および最大立ち
上がり速度については 3000 nmol/L まで作用は認められず、APD50 および APD90 について
3000 nmol/L で適用前値の 114 ± 6%および 116 ± 6%(各 n=6)に延長した。無影響濃度は 300 nmol/L
であった。
b) 麻酔イヌを用いた試験(in vivo)(2.6.3.4 (2) 2) b)、4.2.1.3-14: 試験、GLP準拠)
イヌを用いてイソフルラン麻酔下で TRK-820 の 0、0.1、1 および 10 μg/kg を静脈内漸増投与し
て、血圧(収縮期血圧、拡張期血圧および平均血圧)、心拍数および心電図(PR 間隔、QRS 時間、
QT 間隔および QTc)を投与前、投与終了直後、投与終了 5、15 および 30 分に測定した。各投与
終了直後および投与終了 30 分に採血して血漿中 TRK-820 濃度を測定した。
- 67 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 60
収縮期血圧、拡張期血圧および平均血圧共に TRK-820 の投与により低下が認められ、平均血圧
では、0.1 μg/kg で 77%(%基準値)、1 μg/kg で 61%、10 μg/kg で 53%まで低下した(表 2.6.2-14)。
心拍数は減少傾向を示し、0.1 μg/kg で 90 ± 6%(平均値 ± 標準偏差、投与後 15 分、n=4)、1 μg/kg
で 87 ± 7%(投与後 5 分、n=4)、10 μg/kg で 79 ± 9%(投与後 5 分、n=4)まで減少した。心電図
では PR 間隔、QRS 時間、QT 間隔および QTc 共に 10 μg/kg でも溶媒投与時と比較して変化は認
められなかった。血漿中 TRK-820 濃度はほぼ用量依存的であり、投与終了直後の血漿中濃度は、
0.1 μg/kg で 39.7 ± 24.2 pg/mL(平均値 ± 標準偏差、n=4)、10 μg/kg で 6010 ± 5140 pg/mL(n=4)
であった。
表 2.6.2-14. TRK-820 の麻酔下イヌにおける平均血圧への影響
TRK-820 投与量
(μg/kg, i.v.) 動物数 基準値
(mmHg)
平均血圧 (%基準値) 投与後時間 (分)
直後 5 15 30 0 4 93 ± 15 100.4 ± 9.3 100.8 ± 13.5 100.5 ± 11.7 103.4 ± 16.0 0.1 4 - 82.0 ± 10.1* 79.4 ± 11.2* 76.9 ± 10.6* 79.6 ± 6.6* 1 4 - 67.7 ± 7.6* 62.2 ± 7.5* 60.6 ± 7.3* 63.4 ± 6.3*
10 4 - 58.8 ± 6.1* 53.6 ± 6.0* 53.1 ± 4.3* 55.6 ± 4.9* 数値は平均値 ± 標準偏差(n=4)を示す。 *p<0.05 vs. 溶媒投与群(Dunnett 型多重比較)
(3) 補足的安全性薬理試験
1) 腎/泌尿器系に及ぼす影響(in vivo)(2.6.3.4 (3) 1)、4.2.1.3-15: 試験、GLP準拠)
SD 系雄性ラットに TRK-820(3、10、30、100、300 および 1000 μg/kg)を経口投与し、直後か
ら 6 時間後まで採尿した。3 μg/kg では、尿量および尿中電解質排泄に影響を及ぼさなかった。
10 μg/kg では、尿量、尿中 K+総排泄量および尿中 Cl-総排泄量には影響を及ぼさなかったが、尿中
Na+総排泄量は 29%減少し、統計学的に有意な影響が認められた。30 μg/kg では、尿量および尿中
電解質排泄に影響を及ぼさなかった。100 μg/kg では、尿量、尿中 K+総排泄量および尿中 Cl-総排
泄量には影響を及ぼさなかったが、尿中 Na+総排泄量は 34%減少し、統計学的に有意な影響が認
められた。300 μg/kgでは、尿中Na+総排泄量および尿中Cl-総排泄量には影響を及ぼさなかったが、
尿量が 292%増加および尿中 K+総排泄量が 52%増加し、統計学的に有意な影響が認められた。
1000 μg/kg では尿量は 408%増加、尿中 Na+総排泄量は 45%減少、尿中 K+総排泄量は 54%増加お
よび尿中 Cl-総排泄量は 38%減少し、統計学的に有意な影響が認められた。
- 68 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 61
2) 自律神経系に及ぼす影響(in vitro)(2.6.3.4 (3) 2)、4.2.1.3-16: 試験、GLP準拠)
Hartley 系雄性モルモットの摘出回腸のアセチルコリン、ヒスタミンおよび塩化バリウム刺激に
よる収縮に対して TRK-820(100、300 および 1000 nmol/L)は影響を及ぼさなかった。
3) 胃腸管系に及ぼす影響(in vivo)(2.6.3.4 (3) 3)、4.2.1.3-17: 試験、GLP非準拠)
ICR 系雄性マウスに TRK-820(100、300、1000 および 3000 μg/kg)を経口投与し、30 分後から
20 分の腸管輸送能に対する影響を評価した。100 および 300 μg/kg では腸管輸送能に影響を及ぼさ
なかった。1000 および 3000 μg/kg では腸管輸送能をそれぞれ 32 ± 8 および 53 ± 5%(平均値 ± 標
準誤差、各 n=8)抑制し、統計学的に有意な影響が認められた(ED50:3470 μg/kg、95%信頼区間:
1620~19400 μg/kg)。なお、モルヒネ 100 mg/kg 経口投与では腸管輸送能は 78 ± 3%(n=8)抑制さ
れた。
2.6.2.5 薬力学的薬物相互作用試験
TRK-820 と抗ヒスタミン薬もしくは睡眠導入薬との中枢抑制作用における薬力学的薬物相互作
用を評価した。
(1) 抗ヒスタミン薬ケトチフェンとの相互作用(in vivo)(2.6.3.5 (1) 1)、4.2.1.4-1:試験)
a) 方法
ICR 系雄性マウスにペントバルビタール(50 mg/kg)を腹腔内投与し、正向反射消失から回復
までの期間を睡眠時間として測定した。ケトチフェン(20 mg/kg)をペントバルビタール投与の
60 分前に経口投与し、TRK-820(10 および 100 μg/kg)をペントバルビタール投与の 30 分前に皮
下投与した。ペントバルビタール誘発睡眠時間に対する延長作用を観察し、ケトチフェンの中枢
抑制作用における TRK-820 の併用による影響を評価した。
b) 成績
TRK-820 の 10 μg/kg を皮下投与したときのペントバルビタール誘発睡眠時間は 28.2 ± 3.2 分(平
均値 ± 標準誤差、n=8)であり、ペントバルビタールのみの睡眠時間(32.1 ± 2.5 分(n=8))と同
程度であった。一方、TRK-820 の 100 μg/kg を皮下投与すると、睡眠時間は 54.6 ± 4.4 分(n=8)
となり、ペントバルビタールのみの睡眠時間と比較して 23 分延長し、統計学的に有意な差が認め
られた。また、ケトチフェンの 20 mg/kg を経口投与するとペントバルビタール誘発睡眠時間は
61.2 ± 8.6 分(n=8)となりペントバルビタールのみと比較して 29 分延長し、統計学的に有意な差
が認められた。単独ではペントバルビタール誘発睡眠時間の延長作用を示さない TRK-820 の
10 μg/kg をケトチフェン(20 mg/kg)と併用投与しても睡眠時間はケトチフェン単独投与群よりも
- 69 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 62
8 分短縮するのみ(52.7 ± 4.2 分(n=8))であり、TRK-820 は、ケトチフェンの中枢抑制作用に影
響しなかった(図 2.6.2-14)。一方、単独でペントバルビタール誘発睡眠時間の延長作用を示す
TRK-820 の 100 μg/kg をケトチフェン(20 mg/kg)と併用投与すると、睡眠時間は統計学的に有意
な差はないものの、ケトチフェン単独投与群よりも 20 分延長した(80.7 ± 6.5 分(n=8))(図
2.6.2-14)。20 分の睡眠延長は、TRK-820 の 100 μg/kg 単独投与によって延長した時間である 23 分
と同程度であることから、中枢抑制作用において TRK-820 とケトチフェンが相加的に作用する可
能性が示唆された。
図 2.6.2-14. 中枢神経抑制作用におけるケトチフェンとの相互作用
ペントバルビタール(50 mg/kg)腹腔内投与による睡眠時間を測定 ケトチフェン経口投与の 30 分後に TRK-820 皮下投与 TRK-820 皮下投与の 30 分後にペントバルビタールを腹腔内投与 TRK-820 非投与群: 生理食塩液(溶媒)を 10 mL/kg の容量で皮下投与 ケトチフェン非投与群: 蒸留水(溶媒)を 10 mL/kg の容量で経口投与 縦軸: 正向反射消失から回復までの時間(平均値 ± 標準誤差) 全群 n=8 #p<0.05(Welch 検定)
(2) 睡眠導入薬ニトラゼパムとの相互作用(in vivo)(2.6.3.5 (1) 2)、4.2.1.4-2: 試験)
a) 方法
ICR 系雄性マウスにペントバルビタール(50 mg/kg)を腹腔内投与し、正向反射消失から回復
までの期間を睡眠時間として測定した。ニトラゼパム(3 mg/kg)をペントバルビタール投与の 30
分前に腹腔内投与した。TRK-820(1、10 および 100 μg/kg)はニトラゼパム投与の直前に皮下投
0 0 10 1000
25
50
75
100
TRK-820 (µg/kg, s.c.)
睡眠時間(
分)
#
ケトチフェン (20 mg/kg, p.o.)- + + +
- 70 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 63
与した。ペントバルビタール誘発睡眠時間に対する延長作用を観察し、ニトラゼパムの中枢抑制
作用における TRK-820 の併用による影響を評価した。
b) 成績
ニトラゼパムの 3 mg/kg を腹腔内投与するとペントバルビタール誘発睡眠時間は 68.5 ± 4.4 分
(平均値 ± 標準誤差、n=10)となり、ペントバルビタールのみの睡眠時間(31.4 ± 3.0 分(n=10))
と比較して 37 分延長し、統計学的に有意な差が認められた。TRK-820 の 1 μg/kg をニトラゼパム
(3 mg/kg)と併用投与してもペントバルビタール誘発睡眠時間はニトラゼパム単独投与群と比較
して 2 分短縮するのみ(66.1 ± 6.2 分(n=10))であり、TRK-820 は、ニトラゼパムの中枢抑制作
用に影響しなかった(図 2.6.2-15)。一方、TRK-820の10および100 μg/kgをニトラゼパム(3 mg/kg)
と併用投与すると、睡眠時間はそれぞれ 91.1 ± 2.8 および 96.8 ± 4.4 分(各 n=10)となり、統計学
的に有意な延長作用が認められた(図 2.6.2-15)。したがって、TRK-820 はニトラゼパムの睡眠作
用を用量依存的に増強する可能性が示唆された。
図 2.6.2-15. 中枢神経抑制作用におけるニトラゼパムとの相互作用
ペントバルビタール(50 mg/kg)腹腔内投与による睡眠時間を測定 TRK-820 皮下投与直後にニトラゼパムを腹腔内投与 ニトラゼパム腹腔内投与の 30 分後にペントバルビタールを腹腔内投与 TRK-820 非投与群: 生理食塩液(溶媒)を 10 mL/kg の容量で皮下投与 ニトラゼパム非投与群: 1.5 w/v% CMC 水溶液(懸濁剤)を 10 mL/kg の容量で腹腔内投与 縦軸: 正向反射消失から回復までの時間(平均値 ± 標準誤差) 全群 n=10 ##p<0.01(t 検定) **p<0.01(Dunnett 型多重比較)
0 0 1 10 1000
20
40
60
80
100
120
TRK-820 (µg/kg, s.c.)
睡眠時間(
分)
##
ニトラゼパム (3 mg/kg, i.p.)- + + +
****
+
- 71 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 64
2.6.2.6 考察及び結論
2.6.2.6.1 効力を裏付ける試験 (1) in vitro
TRK-820 のげっ歯類オピオイド受容体選択性は、マウス摘出輸精管標本およびモルモット摘出
回腸標本の電気刺激収縮を指標にした受容体選択性試験により評価し、マウスではオピオイド κ
受容体に対する選択性がオピオイド μおよび δ受容体に対する選択性と比較して高いこと、また、
モルモットにおいてもオピオイド κ 受容体に対する選択性がオピオイド μ 受容体に対する選択性
と比較して高いことが示された。
また、TRK-820 のヒトオピオイド受容体に対する選択性は、オピオイド受容体に対する結合性
試験および作動性試験により検討した。
ヒトオピオイド κ、μ および δ 受容体に対する結合性試験では、TRK-820 のオピオイド κ 受容体
に対する結合性は、オピオイド μ および δ 受容体と比較して、それぞれ 9 および 1984 倍強いこと
が示された。
TRK-820 は、ヒトオピオイド κ 受容体に対しては標準的な完全作動薬 U-69593 と同等の Imaxを
示し、さらに EC50は、U-69593 の約 1/80 および比較的オピオイド κ 受容体に選択的であることが
知られているブトルファノールの約 1/90 であることから、高活性のオピオイド κ 受容体完全作動
薬であることが明らかとなった。一方、オピオイド μ 受容体に対しては標準的な完全作動薬
DAMGO および麻薬鎮痛薬モルヒネと比較して EC50 では、TRK-820 はそれぞれ、1/3.4 および 1/21
であり、低濃度で作用が認められたが、Imaxでは統計学的に有意に低かった。さらにオピオイド μ
受容体の部分作動薬であるブプレノルフィンと同等であることから、TRK-820 はオピオイド μ 受
容体に対して部分作動性を示すと考えられた。オピオイド δ 受容体に対しては標準的な完全作動
薬 DPDPE およびブトルファノールと比較して Imax が統計学的に有意に低く、ブプレノルフィンと
同等であるが、EC50 は DPDPE、ブプレノルフィンおよびブトルファノールのそれぞれ 115、9 お
よび 4 倍高いことから、TRK-820 のオピオイド δ 受容体作動性は弱いと考えられた。
また、オピオイド κ、μ および δ 受容体に対する作動性について、各薬剤の EC50の比は、モル
ヒネで 1:0.1:1.0、ブプレノルフィンで 1:0.4:0.6、ブトルファノールで 1:4.4:6.5 および TRK-820
で 1:203:2610 であった。
したがって、TRK-820 は、オピオイド κ 受容体に対する結合性が強く、さらに、モルヒネ、ブ
プレノルフィンおよびブトルファノールと明確に異なるプロファイルを有し、オピオイド κ 受容
体に対する作動性が非常に強い、オピオイド κ 受容体選択的完全作動薬であることが示唆された。
- 72 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 65
(2) in vivo
各種起痒剤をマウスの吻側背部皮内に投与することによって誘発される引っ掻き行動に対する
TRK-820 の作用について検討した。抗ヒスタミン薬が有効な引っ掻き行動(起痒剤:ヒスタミン)
において、TRK-820 は、30 μg/kg 以上の経口投与で、抗ヒスタミン薬(ケトチフェンおよびクロ
ルフェニラミン)と同様に引っ掻き行動を抑制した。さらに、抗ヒスタミン薬抵抗性の引っ掻き
行動(起痒剤:サブスタンス P およびデオキシコール酸)に対して、TRK-820 はそれぞれ、100
もしくは 30 μg/kg以上の経口投与で有意な抑制作用を示し、ED50はそれぞれ 19.6および 7.62 μg/kg
であった。デオキシコール酸誘発引っ掻き行動モデルにおいて、臨床的に難治性の痒みに有効で
あることが報告されているナルトレキソンは引っ掻き行動を抑制し、ED50は 0.173 mg/kg であっ
たが、抑制率は約 60~70%で頭打ちが認められたのに対して、TRK-820 は、100 μg/kg で完全に引
っ掻き行動を抑制した。また、モルヒネを大槽内投与することよって誘発される抗ヒスタミン薬
抵抗性の中枢性の引っ掻き行動が TRK-820 の 5 および 10 μg/kg の皮下投与で統計学的に有意に抑
制されたことから、TRK-820 は中枢神経系のオピオイド μ 受容体の活性化に伴う痒みに対して止
痒作用を有することが示唆された。さらに、自然発症性のアトピー性皮膚炎モデルマウスの抗ヒ
スタミン薬で抑制し難い引っ掻き行動に対しても、TRK-820 は 100 μg/kg の経口投与で抑制作用を
示し、ED50は 46.1 μg/kg であった。したがって、TRK-820 は、抗ヒスタミン薬が有効な痒みおよ
び抗ヒスタミン薬抵抗性の痒みに対して、広く止痒作用を示す可能性が示唆された。
肝炎、肝硬変、閉塞性黄疸などの慢性肝疾患患者では、しばしば胆汁の流出が停滞する「胆汁
うっ滞」を呈し、全身性の強い痒みが生じることがある 15-17)。また、特定疾患に指定されている
PBC はそう痒を主訴とし、患者の活動性や睡眠を著しく阻害することもある 18)。これらの慢性肝
疾患におけるそう痒症には複数の因子が関与していると考えられている。例えば、痒みを有する
慢性肝疾患患者において血清中のサブスタンス P が上昇していること 19)から、肝疾患患者の痒み
にサブスタンス P が関与していることが示唆されている。一方、慢性肝疾患患者では、血漿中の
Leu-エンケファリンおよび Met-エンケファリン(オピオイド μ 受容体作動性を有する内因性オピ
オイドペプチド)濃度が高いことが報告されている 3, 4)。また、痒みを有する PBC 患者では、痒
みを有さない PBC 患者と比較して、血漿中の β-エンドルフィンおよびエンドモルフィン-1(オピ
オイド μ 受容体作動性を有する内因性オピオイドペプチド)濃度が高いことも報告されている 5)。
さらに、胆汁うっ滞の患者のそう痒に対してオピオイド μ 受容体拮抗薬のナロキソン 6)もしくは
ナルトレキソン 7, 8)が有効であるとの報告もある。加えて、痒みを有する胆汁うっ滞の患者の血漿
抽出物をサルの延髄後角に微量注入すると顔面への引っ掻き行動が誘発され、その引っ掻き行動
はナロキソンにより抑制されるとの報告もあること 20)から、肝疾患患者における痒みの発現には
オピオイド μ 受容体の活性化が関与していることが示唆されている。したがって、TRK-820 がサ
ブスタンス P 皮内投与誘発マウス引っ掻き行動およびモルヒネ大槽内投与誘発マウス引っ掻き行
- 73 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 66
動を抑制したことから、TRK-820 が肝疾患患者の痒みに対して十分な有効性を示す可能性がある
ものと考えられた。
また、デオキシコール酸などの胆汁酸をヒトの皮膚に実験的に誘発した水疱の底部に適用する
と痒みを引き起こすこと 21)から、胆汁中に含まれる胆汁酸が胆汁うっ滞性の痒みに関与している
ことが示唆されている。TRK-820 はデオキシコール酸の皮内投与により誘発されるマウスの引っ
掻き行動を抑制したことから、胆汁うっ滞性のそう痒に対して有効である可能性が考えられた。
なお、肝疾患に伴う痒みのモデルとしては、デオキシコール酸皮内投与誘発マウス引っ掻き行
動モデルのほか、エチニルエストラジオール反復投与誘発ラット引っ掻き行動モデル(胆汁うっ
滞性のそう痒モデル)22)および MRL/lpr 系マウスの引っ掻き行動モデル(PBC に伴う痒みのモデ
ル)23)が報告されている。しかし、エチニルエストラジオール反復投与モデルにおいてはラット
では引っ掻き行動が認められるが、マウスでは引っ掻き行動が認められないと報告されているこ
と 22)、TRK-820 の経口投与によるバイオアベイラビリティが、マウス(32%)と比較してラット
(4.6%)で非常に低いこと、また、MRL/lpr 系マウスは PBC だけではなく、全身性エリテマトー
デス、尋常性天疱瘡、シェーグレン・ラルソン症候群もしくは多発性硬化症などのその他の痒み
を伴う自己免疫疾患のモデルでもあること 23)から、肝疾患に伴う痒みのモデルとして、デオキシ
コール酸皮内投与誘発マウス引っ掻き行動モデルを選択し、実施した。なお、TRK-820 はエチニ
ルエストラジオール反復投与誘発ラット引っ掻き行動 22)および MRL/lpr 系マウスの引っ掻き行動
23)のいずれに対しても抑制作用を示すことが報告されており、胆汁うっ滞および自己免疫疾患に
おける痒みに対して有効である可能性が示唆されている。
さらに、抗ヒスタミン薬抵抗性の自然発症アトピー性皮膚炎モデルである NC/Nga 系マウス 11)
の引っ掻き行動を TRK-820 が抑制したことから、TRK-820 は抗ヒスタミン薬の効果が不十分であ
るといわれているアトピー性皮膚炎に伴う痒みに対しても有効である可能性が示唆された。
TRK-820(100 μg/kg)を経口投与した際の引っ掻き行動抑制作用の持続時間を評価したところ、
投与 6 時間後まではサブスタンス P 誘発引っ掻き行動を有意に抑制したが、投与 8 時間後には有
意な抑制作用は消失した。したがって、マウスに TRK-820 の 100 μg/kg を経口投与したときの止
痒作用持続時間は、約 6 時間であることが示された。[3H]TRK-820 をマウスに単回経口投与
(100 μg/kg)した 6 および 24 時間後における大脳内放射能濃度が、それぞれ 0.40 および
0.11 ng eq. of TRK-820/g(0.78 および 0.21 nmol eq. of TRK-820/kg)であり(2.6.4.4、表 2.6.4.4-1)、
TRK-820 のマウス摘出輸精管電気刺激誘発収縮に対する抑制作用(主としてオピオイド κ 受容体
を介する作用)の IC50(0.080~0.12 nmol/L)以上の濃度が、マウスの脳内に長時間存在している
ことを考慮すると、TRK-820 のマウスにおける止痒作用が約 6 時間であったことは妥当な結果で
あると考えられた。
- 74 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 67
止痒作用が認められる用量の TRK-820(100 μg/kg)もしくは溶媒を 1 日 2 回、7 日間反復経口
投与することによって溶媒反復投与群では TRK-820 の 25 μg/kg 以上、TRK-820 反復投与群では
100 μg/kg 以上で統計学的に有意な引っ掻き行動抑制作用が認められた。したがって、TRK-820 の
反復経口投与により引っ掻き行動抑制作用は減弱することが示唆された。モルヒネの鎮痛作用に
対する耐性形成に関する文献では、鎮痛作用が認められる用量のモルヒネを 1 日 1 回、6 日間 13)
もしくは 1 日 2 回、9 日間 14)反復投与すると鎮痛作用がほぼ完全に消失することが報告されてい
る。一方、TRK-820 の止痒作用が認められる用量(100 μg/kg)の反復投与によって、同用量の
TRK-820 の引っ掻き行動抑制作用は消失していないこと、引っ掻き行動に対する ED50は、1.8 倍
増加したのみであったことから、TRK-820 は止痒作用において強い耐性を形成する可能性は低い
と考えられた。
(3) 作用機序
炎症反応として肥満細胞から遊離されるヒスタミンは痒みの主要なメディエーターとして広く
知られている。さらに、白血球などから分泌されるサイトカイン(IL-2、IL-6 など)が痒みに関
連する可能性 24, 25)や、ケラチノサイトが産生する NO が痒みの増強因子として働く可能性 26)が報
告されている。また、結膜の痒みにプロスタグランジン類(PGE2、PGI2 および PGD2)が関与す
る可能性も報告されている 27)。しかしながら、TRK-820 は、in vitro において炎症性メディエータ
ー遊離(ヒスタミン遊離、TNF-α 分泌、IL-1β 分泌、IL-6 分泌、PGE2 分泌および PGD2分泌)お
よび NOS 活性(誘導型 NOS および構成型 NOS)に対して阻害作用を示さなかった。また、TRK-820
は、ムスカリン M1受容体に対する[3H]ピレンゼピンの結合を 1000 および 10000 nmol/L でそれぞ
れ 41 および 72%阻害したが、その Ki 値は 1700 nmol/L であった。さらに、TRK-820 は 1000 nmol/L
でオルファニン ORL1 受容体に対する[3H]ノシセプチンの結合を 47%阻害した。加えて、TRK-820
は 1000 もしくは 10000 nmol/L でアデノシン A2A受容体、アドレナリン α1受容体、アドレナリン α2
受容体、ボンベシン受容体、CGRP 受容体、ドパミン D2受容体、GABAA受容体 Cl-チャネル、グ
ルタミン酸 mGluR5受容体、グルタミン酸 NMDA 受容体(Agonist Site)、オレキシン OX1受容体、
セロトニン 5-HT2受容体、セロトニン 5-HT4 受容体およびシグマ受容体に対する各特異的標識リ
ガンドの結合を 10~27%阻害した。したがって、TRK-820 は上記の受容体に対して低い結合性を
示したが、オピオイド κ 受容体に対する結合性と比較すると著しく低かった。その他の受容体、
イオンチャネルおよびトランスポーターに対する各特異的標識リガンドの結合に対する TRK-820
の 1000 または 10000 nmol/L での阻害率は、10%未満であり、結合性をほとんど示さなかった。
一方、in vivo において TRK-820(100 μg/kg)経口投与によるサブスタンス P 皮内投与誘発マウ
ス引っ掻き行動の抑制作用は、オピオイド κ 受容体拮抗薬 nor-BNI の皮下投与(10 mg/kg)によっ
て部分的に拮抗され、TRK-820(10 μg/kg)皮下投与による作用は、nor-BNI の脳室内投与(10 μg/site)
- 75 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 68
によって完全に消失した。また、TRK-820(0.01~10 μg/mL)は、モルモットにおいて局所麻酔作
用を示さなかった。また、オピオイド受容体に対する結合試験および作動性試験の結果から、
TRK-820 はオピオイド κ 受容体選択的完全作動薬であることが示唆されている。以上のことから、
TRK-820 の引っ掻き行動抑制作用は、オピオイド κ 受容体、特に中枢神経系のオピオイド κ 受容
体の活性化を介して発現していることが示唆された。
また、オピオイド κ 受容体は、一般的にオピオイド μ 受容体と相反する作用を有することが知
られており 28)、TRK-820 がモルヒネ大槽内投与誘発マウス引っ掻き行動を抑制したことから、
TRK-820 は中枢神経系のオピオイド μ 受容体活性化に伴う痒みのシグナルを抑制することで止痒
作用を発現することも示唆された。
TRK-820 経口剤中の不純物である 10α-OH は、in vitro 受容体結合試験において 10000 nmol/L で
ヒトオピオイド κ、μ および δ 受容体に対する特異的リガンド結合をそれぞれ 100、66 および 8%
阻害したが、オピオイド κ および μ 受容体に対する Ki 値は、それぞれ 4.26 および 2070 nmol/L で
あり、TRK-820 と比較してそれぞれ 17 および 937 倍高値であった。さらに、10α-OH は、in vitro
受容体作動性試験において、オピオイド κ 受容体に対して完全作動性を示したものの、EC50は
TRK-820 よりも 6.9 倍高濃度であった。また、10α-OH は、オピオイド μ および δ 受容体に対して
は TRK-820 よりも極めて弱い作動性を示したのみであり、EC50 を算出することができなかった。
TRK-820 経口剤中の 10α-OH の規格は %以下と設定されている(2.3.P.5.1)ことから、経口剤中
の 10α-OHはオピオイド受容体に対する作用を発現せず、止痒作用にも関与しないと考えられた。
TRK-820 の代謝物である de-CPM、NFA-G および de-CPM-G のヒトオピオイド受容体に対する
結合性を in vitro にて評価したところ、de-CPM の 10000 nmol/L でのオピオイド κ、μ および δ 受
容体に対する特異的リガンド結合の阻害率は、それぞれ 103、93 および 25%であり、NFA-G の
10000 nmol/L でのオピオイド κ、μ および δ 受容体に対する特異的リガンド結合の阻害率は、それ
ぞれ 73、25 および 3%であった。de-CPM-G の 10000 nmol/L でのオピオイド κ、μ および δ 受容体
に対する特異的リガンド結合の阻害率は、それぞれ 6、-1 および-8%であった。また、オピオイド
κ 受容体に対する de-CPM の Ki 値は 5.95 nmol/Lであり、TRK-820 と比較して 24 倍高値であった。
オピオイド μ 受容体に対する de-CPM の Ki 値は 133 nmol/L であり、TRK-820 と比較して 60 倍高
値であった。また、オピオイド κ 受容体に対する NFA-G の Ki 値は 1960 nmol/L であり、TRK-820
と比較して 8033 倍高値であった。さらに in vitro 受容体作動性試験において、オピオイド κ 受容
体に対して de-CPM および NFA-G は、完全作動性を示したものの、EC50は TRK-820 よりもそれ
ぞれ 272 および 4600 倍高濃度であり、de-CPM-G は試験で使用した最高濃度の 3000 nmol/L でも
TRK-820 の 24%程度の作動性を示したのみであった。また、オピオイド μ および δ 受容体に対し
ては、いずれの代謝物も TRK-820 と比較して極めて弱い作動性を示したのみであり、EC50 を算出
することはできなかった。また、de-CPM、NFA-G および de-CPM-G のオピオイド受容体以外の受
- 76 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 69
容体、イオンチャネルおよびトランスポーターに対する結合性を in vitro にて 1000 nmol/L で評価
したところ、各代謝物が特異的標識リガンドの結合に対して最大阻害率を示した受容体は、
de-CPM ではセロトニン 5-HT4 受容体(阻害率:23%)、NFA-G および de-CPM-G ではプリン P2Y
受容体(阻害率:それぞれ 22 および 31%)であり、いずれも TRK-820 のオピオイド κ 受容体に
対する結合性と比較すると著しく低かった。その他の受容体、イオンチャネルおよびトランスポ
ーターの各特異的標識リガンド結合に対する de-CPM、NFA-G および de-CPM-G の阻害率は上記
よりも低く、結合性をほとんど示さなかった。
これに加えて、サブスタンス P 皮内投与誘発マウス引っ掻き行動に対して、de-CPM·T、NFA-G
および de-CPM-G を、TRK-820 が皮下投与により統計学的に有意な抑制作用を示す用量(10 μg/kg)
の 100 倍量(1000 μg/kg)まで皮下投与しても抑制作用を発現しなかったことから、de-CPM、NFA-G
および de-CPM-G は TRK-820 の止痒作用発現に関与しないと考えられた。
以上のことから、TRK-820 の止痒作用は、未変化体による中枢神経系のオピオイド κ 受容体の
活性化に起因することが示唆された。
2.6.2.6.2 安全性薬理試験
(1) 中枢神経系に及ぼす影響(in vivo)
安全性薬理コアバッテリー試験において経口投与した TRK-820 は、ラットの一般症状および行
動に対して 1000 μg/kg では身づくろいの軽度な低下、自発運動の軽度な低下、体温の軽度な低下
および軽度な眼瞼下垂、3000 μg/kg では警戒性の軽度な低下、身づくろいの軽度な低下、反応性
の軽度な低下、自発運動の軽度な低下、軽度なよろめき歩行、軽度な眼瞼下垂、体温の軽度な低
下ないし低下、軽度な四肢伸長、軽度な流涙、逃避反応の軽度な低下、痛覚反応の軽度な低下、
耳介反射の軽度な低下、軽度な腹這い姿勢、軽度な異常歩行および軽度な縮瞳を引き起こした。
したがって、ラットにおいて主に中枢神経系の抑制作用に基づくと考えられる一般症状の変化が
認められた。
さらに、安全性薬理コアバッテリーに対するフォローアップ試験において以下を確認した。
静脈内投与した TRK-820 は、アカゲザルに対して 0.5 μg/kg では 2 例中 2 例で運動低下、うずく
まり姿勢および観察者への攻撃行動の増強、同 2 例中 1 例で観察者への攻撃行動の減弱、腹臥位
および動作緩慢、1 μg/kg では 2 例中 2 例で観察者への攻撃行動の減弱、同 2 例中 1 例で運動低下、
うずくまり姿勢、腹臥位、観察者への怯え表情の減弱、動作緩慢および閉眼、2 μg/kg では 2 例中
2 例で流涎、運動低下、うずくまり姿勢、閉眼、動作緩慢および口の半開状態、同 2 例中 1 例で
観察者への攻撃行動の減弱、腹臥位および運動失調を引き起こした。
以上のように、サルにおいても、安全性薬理コアバッテリー試験の Irwin 法によるラットの一般
症状観察に見られたのと同様に、主に中枢神経系の抑制作用に基づくと考えられる一般症状の変
- 77 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 70
化が認められた。アカゲザルにおけるモルヒネ静脈内投与誘発引っ掻き行動が TRK-820 の
0.25 μg/kg 静脈内投与で統計学的に有意に抑制されることが報告されている 29)ことから、一般症
状の変化は止痒作用が認められる用量よりも高用量で発現することが考えられた。
経口投与した TRK-820 は、マウスの移所運動を 400 μg/kg で統計学的に有意に減少させ、ED50
は 345 μg/kg であった。各種起痒剤の皮内投与によって誘発されるマウス引っ掻き行動に対する
TRK-820 の抑制作用の ED50 は、7~20 μg/kg(経口投与)であることから、自発運動抑制作用は止
痒作用が認められる用量の 17~49 倍高用量で発現することが示唆された。さらに、TRK-820 はラ
ットの移所運動を 1000 および 3000 μg/kg の経口投与で統計学的に有意に減少させた。
なお、移所運動よりも自発運動抑制作用を検出しやすいといわれている回転かご試験では、マ
ウスの自発運動は、TRK-820 の 100 および 300 μg/kg 経口投与で統計学的に有意に抑制された。
ED50は 103(30 分間測定)もしくは 90.6(60 分間測定)μg/kg であり、止痒作用が認められる用
量(ED50:7~20 μg/kg、経口投与)の 5~15 倍高用量で自発運動抑制作用が発現することが示唆
された。なお、TRK-820 の 10 および 30 μg/kg 皮下投与で統計学的に有意な自発運動の抑制作用が
認められ、ED50は 7.79 μg/kg であった。
経口投与した TRK-820 は、マウスのロタロッド試験において 30 分後では 300 μg/kg で統計学的
に有意に協調運動を阻害し、ED50は 115 μg/kg であった。60 分後では、100 および 300 μg/kg で統
計学的に有意に協調運動を阻害し、ED50は 72.3 μg/kg であった。各種起痒剤の皮内投与によって
誘発されるマウス引っ掻き行動に対する TRK-820 の抑制作用(ED50:7~20 μg/kg)は TRK-820
経口投与後 30 分後に評価していることから、TRK-820 は止痒作用が認められる用量の 6~16 倍高
用量で協調運動を阻害することが考えられた。
経口投与した TRK-820 は、ペントバルビタール(50 mg/kg)の腹腔内投与によるマウスの睡眠
時間に対して 1000 μg/kg 以上の用量で、統計学的に有意な延長作用を発現した。各種起痒剤の皮
内投与によって誘発されるマウス引っ掻き行動に対して、TRK-820 は 30 もしくは 100 μg/kg の経
口投与により統計学的に有意な抑制作用を発現することから、止痒作用が認められる用量の 10 も
しくは 30 倍高用量で麻酔作用を発現することが考えられた。
皮下投与した TRK-820 は、ラットの新皮質前頭葉の脳波の周波数解析に対して、3 μg/kg でアル
ファ帯域の減少、10 および 30 μg/kg でアルファおよびベータ-1 帯域の減少、海馬の周波数解析に
対しては、10 μg/kg でアルファ帯域の減少およびベータ-2 帯域の増加、30 μg/kg でアルファ帯域
の減少を引き起こした。睡眠-覚醒周期の各脳波水準に対しては、3 μg/kg で覚醒期の増加、徐波睡
眠期の減少および速波睡眠期の減少、10 μg/kg で覚醒期の増加、徐波睡眠期の減少、速波睡眠期
の減少および速波睡眠期の潜時時間の延長、30 μg/kg で覚醒期の増加、徐波睡眠期の減少、速波
睡眠期の減少、徐波睡眠期の潜時時間の延長および速波睡眠期の潜時時間の延長を引き起こした。
予備的な結果であるが、TRK-820 の皮下投与により、ヒスタミン皮内投与誘発ラット引っ掻き行
- 78 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 71
動が 3 μg/kg 以上で有意に抑制される(4.2.1.1-39: 試験)ことから、TRK-820 は止痒作
用が認められる用量とほぼ同じ用量で覚醒期の増加を引き起こすことが示唆された。
経口投与した TRK-820 は、マウス酢酸ライジング試験による鎮痛試験において 25 μg/kg 以上で
統計学的に有意なライジング回数の減少が観察され、ED50は 31.9 μg/kg であった。また、TRK-820
の皮下投与によるラット Paw pressure およびホルマリン鎮痛試験における ED50 は、それぞれ 64
および 9.6 μg/kg であると報告されている 30)。したがって、各種起痒剤の皮内投与によって誘発さ
れるマウス引っ掻き行動に対する TRK-820 の抑制作用(ED50:7~20 μg/kg、経口投与)もしくは
ヒスタミン皮内投与誘発ラット引っ掻き行動に対する TRK-820 の抑制作用(ED50:1.25 μg/kg、皮
下投与(4.2.1.1-39: 試験))と比較すると、TRK-820 は止痒作用が認められる用量より
も高用量で鎮痛作用を発現することが考えられた。
経口投与した TRK-820 は、ラットの体温に対して 1000 μg/kg では溶媒対照群と比較して 0.3~
0.5°C の軽度な体温低下、3000 μg/kg では 1.2~1.5°C の体温低下を引き起こしたことから、比較的
高用量で体温低下作用を発現することが考えられた。
(2) 心血管系に及ぼす影響
安全性薬理コアバッテリー試験として実施した hERG 電流に対する作用に関する試験において、
hERG 遺伝子を導入した HEK-293 細胞におけるカリウム電流(IKr)に対する TRK-820 の IC50 は
840 nmol/L であり、無作用量は 3 nmol/L であった。
安全性薬理コアバッテリーに対するフォローアップ試験として実施したモルモット摘出乳頭筋
の活動電位に対する作用に関する試験において、TRK-820 は、3000 nmol/L で APD50 および APD90
をそれぞれ適用前値の 114%および 116%に延長した。無作用量は 300 nmol/L であった。
hERG電流のIC50 840 nmol/L(431 ng/mL)およびモルモット摘出乳頭筋の活動電位に対する無作
用量 300 nmol/L(154 ng/mL)は、Child-Pugh分類グレードAの代償性肝硬変患者を対象とした臨床
薬理試験(5.3.3.2-1:820CPC01 試験)における単回経口投与時のデータから推定した予想臨床使
用最高用量 5 μg/bodyの反復経口投与時のCmax 6.79 pg/mL(2.7.2.3.2 (1)、表 2.7.2-22)と比較して、
それぞれ約 63000 および 23000 倍高濃度であった。また、Child-Pugh分類グレードBの肝硬変患者
を対象とした臨床薬理試験(5.3.3.2-2:820HPC02 試験)における単回経口投与時のデータから推
定した予想臨床使用最高用量 5 μg/bodyの反復経口投与時のCmax 14.8 pg/mL(2.7.2.3.2 (1)、表
2.7.2-23)と比較して、それぞれ約 29000 および 10000 倍高濃度であった。また、安全性薬理コア
バッテリー試験として実施した無麻酔・非拘束イヌを用いた試験において、TRK-820 を経口投与
したときのQTcに対する無作用量は 300 μg/kgより大きいと考えられ、血圧、心拍数を含めた心血
管系に対する無作用量は 3 μg/kgであった。イヌに 3 および 300 μg/kgを経口投与したときのCmax
- 79 -
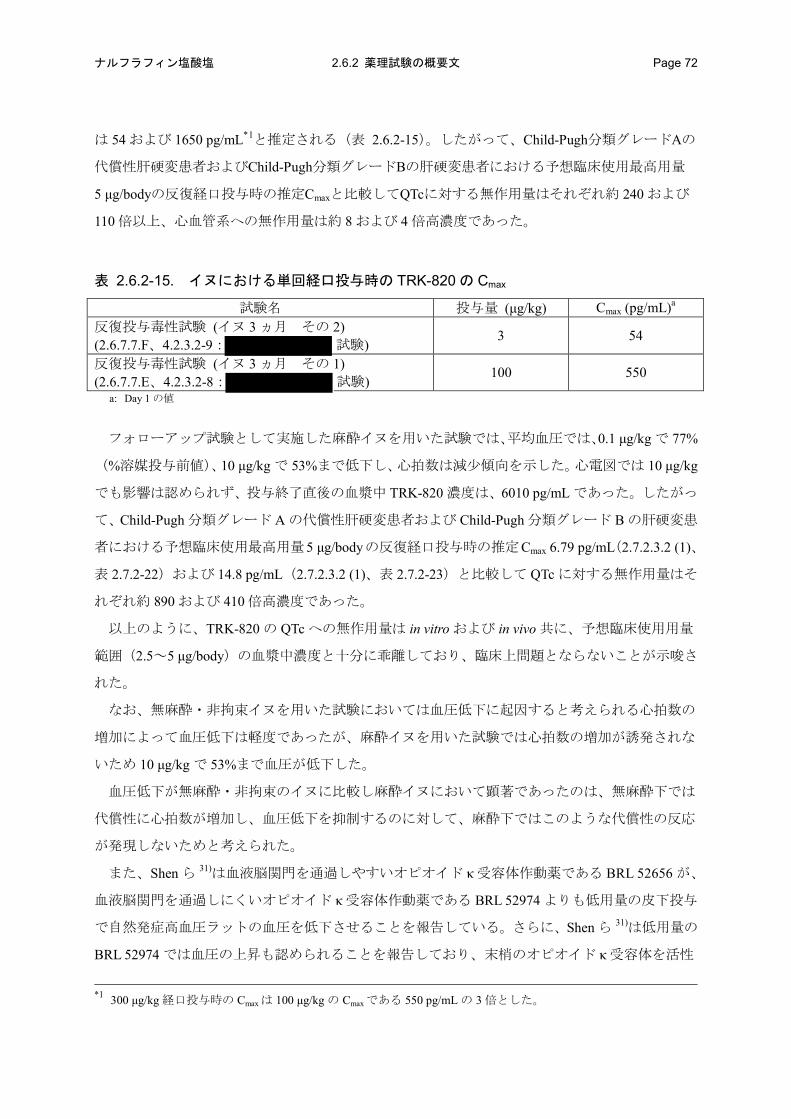
ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 72
は 54 および 1650 pg/mL*1と推定される(表 2.6.2-15)。したがって、Child-Pugh分類グレードAの
代償性肝硬変患者およびChild-Pugh分類グレードBの肝硬変患者における予想臨床使用最高用量
5 μg/bodyの反復経口投与時の推定Cmaxと比較してQTcに対する無作用量はそれぞれ約 240 および
110 倍以上、心血管系への無作用量は約 8 および 4 倍高濃度であった。
表 2.6.2-15. イヌにおける単回経口投与時の TRK-820 の Cmax
試験名 投与量 (μg/kg) Cmax (pg/mL)a 反復投与毒性試験 (イヌ 3 ヵ月 その 2) (2.6.7.7.F、4.2.3.2-9: 試験)
3 54
反復投与毒性試験 (イヌ 3 ヵ月 その 1) (2.6.7.7.E、4.2.3.2-8: 試験)
100 550
a: Day 1 の値
フォローアップ試験として実施した麻酔イヌを用いた試験では、平均血圧では、0.1 μg/kg で 77%
(%溶媒投与前値)、10 μg/kg で 53%まで低下し、心拍数は減少傾向を示した。心電図では 10 μg/kg
でも影響は認められず、投与終了直後の血漿中 TRK-820 濃度は、6010 pg/mL であった。したがっ
て、Child-Pugh 分類グレード A の代償性肝硬変患者および Child-Pugh 分類グレード B の肝硬変患
者における予想臨床使用最高用量 5 μg/bodyの反復経口投与時の推定Cmax 6.79 pg/mL(2.7.2.3.2 (1)、
表 2.7.2-22)および 14.8 pg/mL(2.7.2.3.2 (1)、表 2.7.2-23)と比較して QTc に対する無作用量はそ
れぞれ約 890 および 410 倍高濃度であった。
以上のように、TRK-820 の QTc への無作用量は in vitro および in vivo 共に、予想臨床使用用量
範囲(2.5~5 μg/body)の血漿中濃度と十分に乖離しており、臨床上問題とならないことが示唆さ
れた。
なお、無麻酔・非拘束イヌを用いた試験においては血圧低下に起因すると考えられる心拍数の
増加によって血圧低下は軽度であったが、麻酔イヌを用いた試験では心拍数の増加が誘発されな
いため 10 μg/kg で 53%まで血圧が低下した。
血圧低下が無麻酔・非拘束のイヌに比較し麻酔イヌにおいて顕著であったのは、無麻酔下では
代償性に心拍数が増加し、血圧低下を抑制するのに対して、麻酔下ではこのような代償性の反応
が発現しないためと考えられた。
また、Shen ら 31)は血液脳関門を通過しやすいオピオイド κ 受容体作動薬である BRL 52656 が、
血液脳関門を通過しにくいオピオイド κ 受容体作動薬である BRL 52974 よりも低用量の皮下投与
で自然発症高血圧ラットの血圧を低下させることを報告している。さらに、Shen ら 31)は低用量の
BRL 52974 では血圧の上昇も認められることを報告しており、末梢のオピオイド κ 受容体を活性
*1 300 μg/kg 経口投与時の Cmax は 100 μg/kg の Cmaxである 550 pg/mL の 3 倍とした。
- 80 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 73
化させると血圧が上昇する可能性が示されている。したがって、オピオイド κ 受容体作動薬の血
圧低下作用は中枢神経系のオピオイド κ 受容体への作用を介して発現している可能性が考えられ
た。また、Wright ら 32)はオピオイド κ 受容体のアンチセンスオリゴヌクレオシドを自然発症高血
圧ラットおよび正常血圧ラットの脳内(海馬)に投与し、オピオイド κ 受容体をノックダウンさ
せることにより血圧が上昇することを報告している。さらに、Rao ら
33)は正常血圧ラットの脳室
内にオピオイド κ 受容体作動性ペプチドであるダイノルフィンを投与することにより血圧低下と
腸間膜動脈の弛緩が認められること、オピオイド κ 受容体拮抗薬の nor-BNI を脳室内投与すると
血圧上昇と腸間膜動脈の収縮が観察されることを報告している。以上のことから、TRK-820 の血
圧低下作用は、中枢神経系のオピオイド κ 受容体の活性化に基づく可能性が考えられた。
(3) 呼吸系に及ぼす影響(in vivo)
無麻酔・非拘束イヌを用いた試験で TRK-820 の経口投与による呼吸系に対する無作用量は
300 μg/kg より大きいと考えられ、推定される Cmax は 1650 pg/mL(表 2.6.2-15)であることから、
Child-Pugh 分類グレード A の代償性肝硬変患者および Child-Pugh 分類グレード B の肝硬変患者に
おける予想臨床使用最高用量 5 μg/body の反復経口投与時の推定 Cmax 6.79 pg/mL(2.7.2.3.2 (1)、表
2.7.2-22)および 14.8 pg/mL(2.7.2.3.2 (1)、表 2.7.2-23)と比較して、呼吸系への無作用量はそれ
ぞれ約 240 および 110 倍以上高濃度であった。
(4) その他の安全性薬理試験
経口投与した TRK-820 は、ラットにおいて 10 μg/kg で、尿中 Na+総排泄量を 29%減少させたが、
30 μg/kg ではその作用が消失した。また、100 μg/kg で、尿中 Na+総排泄量を 34%減少させ、300 μg/kg
で、尿量を 292%増加および尿中 K+総排泄量を 52%増加させ、1000 μg/kg で、尿量を 408%増加、
尿中 Na+総排泄量を 45%減少、尿中 K+総排泄量を 54%増加および尿中 Cl-総排泄量を 38%減少さ
せた。この変動については、ラット 6 ヵ月間反復経口投与毒性試験(2.6.7.7.C、4.2.3.2-3:
)、イヌ 3 ヵ月間反復経口投与毒性試験(2.6.7.7.E、4.2.3.2-8: 試験)
などにおいて、血清中電解質濃度および病理組織学的検査で腎臓に異常所見が見られなかったこ
とから、毒性学的意義は低いと考えられた。
経口投与した TRK-820 は、マウス腸管輸送能試験において 1000 および 3000 μg/kg で、それぞ
れ 32 および 53%の抑制作用を示したのみであり、ED50 は 3470 μg/kg となった。したがって、腸
管輸送能抑制作用は止痒作用の 177~475 倍高用量で発現し、その作用はモルヒネ(100 mg/kg 経
口投与時の抑制率:78%)と比較して弱いことが明らかとなった。
- 81 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 74
2.6.2.6.3 薬力学的薬物相互作用試験
ペントバルビタール(50 mg/kg)の腹腔内投与によって誘発されるマウスの睡眠時間に対する
抗ヒスタミン薬ケトチフェン(20 mg/kg)経口投与による延長作用は、単独投与でペントバルビ
タール誘発睡眠時間に影響のない TRK-820 の 10 μg/kg を皮下投与しても影響を受けなかった。一
方、単独投与でペントバルビタール誘発睡眠時間を延長する TRK-820 の 100 μg/kg の皮下投与に
よってケトチフェンの睡眠時間延長作用は統計学的に有意ではないものの、20 分延長した。一方、
睡眠導入薬ニトラゼパム(3 mg/kg)腹腔内投与によるペントバルビタール誘発睡眠時間の延長作
用は、TRK-820 の 1 μg/kg 皮下投与では影響を受けなかったが、TRK-820 の 10 および 100 μg/kg
では統計学的に有意に延長された。したがって、TRK-820 と抗ヒスタミン薬は中枢抑制作用にお
いて相加的に作用し、また、TRK-820 が睡眠導入薬の睡眠作用を用量依存的に増強する可能性が
示唆された。
なお、TRK-820 はサブスタンス P 誘発マウス引っ掻き行動を 10 μg/kg の皮下投与で統計学的に
有意に抑制するのに対して、ケトチフェンの睡眠時間延長作用は TRK-820 の 10 μg/kg の皮下投与
では影響を受けず、100 μg/kg の皮下投与により 20 分延長したことから、抗ヒスタミン薬との併
用では、止痒作用が認められる用量よりも 10 倍高用量で中枢抑制作用において相加的な作用が発
現することが考えられた。したがって、TRK-820 と抗ヒスタミン薬との相互作用については臨床
使用上の問題はないと考えられた。一方、睡眠導入薬との併用では、サブスタンス P 誘発マウス
引っ掻き行動を統計学的に有意に抑制する比較的低用量の TRK-820(10 μg/kg)皮下投与で既に相
互作用が発現した。したがって、TRK-820 の臨床使用上において睡眠導入薬との併用には注意す
る必要があると考えられた。
以上の非臨床薬理試験の結果から、TRK-820 は抗ヒスタミン薬などの既存薬とは異なり、中枢
神経系のオピオイド κ 受容体の活性化という新規なメカニズムを介して、難治性の痒みに対して
優れた止痒作用を発現する可能性が示唆された。さらに、安全性薬理試験の結果から、予想臨床
使用最高用量の Cmaxにおいて臨床使用上の大きな問題はないと考えられた。したがって、TRK-820
は「慢性肝疾患患者における難治性のそう痒症に対する止痒薬」として優れた有効性および安全
性が期待できると考えられた。
2.6.2.7 図表
本文中に記載。
- 82 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 75
2.6.2.8 参考文献
1) Gillespie DA, Vickers CR. Pruritus and cholestasis: therapeutic options. J Gastroenterol Hepatol. 1993;8:168-73. 【4.3-14】
2) Mela M, Mancuso A, Burroughs AK. Review article: pruritus in cholestatic and other liver diseases. Aliment Pharmacol Ther. 2003;17:857-70. 【4.3-15】
3) Thornton JR, Losowsky MS. Plasma leucine enkephalin is increased in liver disease. Gut. 1989;30:1392-5. 【4.3-17】
4) Thornton JR, Losowsky MS. Opioid peptides and primary biliary cirrhosis. BMJ. 1988;297:1501-4. 【4.3-18】
5) 川島由美. 原発性胆汁性肝硬変におけるそう痒の原因物質に関する研究―opioid peptide の関
与―. 帝京医学雑誌. 2005;28:89-97. 【4.3-19】
6) Bergasa NV, Alling DW, Talbot TL, Swain MG, Yurdaydin C, Turner ML, et al. Effects of naloxone infusions in patients with the pruritus of cholestasis. A double-blind, randomized, controlled trial. Ann Intern Med. 1995;123:161-7. 【4.3-20】
7) Terg R, Coronel E, Sordá J, Muñoz AE, Findor J. Efficacy and safety of oral naltrexone treatment for pruritus of cholestasis, a crossover, double blind, placebo-controlled study. J Hepatol. 2002;37:717-22. 【4.3-5】
8) Wolfhagen FHJ, Sternieri E, Hop WCJ, Vitale G, Bertolotti M, Van Buuren HR. Oral naltrexone treatment for cholestatic pruritus: a double-blind, placebo-controlled study. Gastroenterology. 1997;113:1264-9. 【4.3-4】
9) Tohda C, Yamaguchi T, Kuraishi Y. Intracisternal injection of opioids induces itch-associated response through μ-opioid receptors in mice. Jpn J Pharmacol. 1997;74:77-82. 【4.3-55】
10) Takano N, Arai I, Kurachi M. Analysis of the spontaneous scratching behavior by NC/Nga mice: a possible approach to evaluate antipruritics for subjects with atopic dermatitis. Eur J Pharmacol. 2003;471:223-8. 【4.3-1】
11) Takano N, Arai I, Hashimoto Y, Kurachi M. Evaluation of antipruritic effects of several agents on scratching behavior by NC/Nga mice. Eur J Pharmacol. 2004;495:159-65. 【4.3-2】
12) 中尾薫, 池田顕, 黒川敬弘, 冨樫裕子, 梅内秀郎, 本多敏行, 他. アトピー性皮膚炎モデルの
引っ掻き行動に対するオピオイド κ受容体作動薬TRK-820の効果. 日本神経精神薬理学雑誌. 2008;28:75-83. 【4.3-3】
13) Tsuji M, Yamazaki M, Takeda H, Matsumiya T, Nagase H, Tseng LF, et al. The novel κ-opioid receptor agonist TRK-820 has no affect on the development of antinociceptive tolerance to morphine in mice. Eur J Pharmacol. 2000;394:91-5. 【4.3-36】
- 83 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 76
14) Raghavendra V, Kulkarni SK. Possible mechanisms of action in melatonin reversal of morphine tolerance and dependence in mice. Eur J Pharmacol. 2000;409:279-89. 【4.3-37】
15) 東田千尋. 肝障害のかゆみとオピオイド. 医学のあゆみ. 2001;197:616-7. 【4.3-10】
16) 伊崎誠一. Q30 肝疾患に伴うかゆみについて. In: 宮地良樹編. かゆみ Q&A. 医薬ジャーナル
社, 1997;76-7. 【4.3-11】
17) Jones EA, Bergasa NV. The pruritus of cholestasis. Hepatology. 1999;29:1003-6. 【4.3-12】
18) Bergasa NV. Pruritus and fatigue in primary biliary cirrhosis. Clin Liver Dis. 2003;7:879-900. 【4.3-13】
19) Trivedi M, Bergasa NV. Serum concentrations of substance P in cholestasis. Ann Hepatol. 2010;9:177-80. 【4.3-16】
20) Bergasa NV, Thomas DA, Vergalla J, Turner ML, Jones EA. Plasma from patients with the pruritus of cholestasis induces opioid receptor-mediated scratching in monkeys. Life Sci. 1993;53:1253-7. 【4.3-21】
21) Kirby J, Heaton KW, Burton JL. Pruritic effect of bile salts. Br Med J. 1974;4:693-5. 【4.3-56】
22) Inan S, Cowan A. Nalfurafine, a kappa opioid receptor agonist, inhibits scratching behavior secondary to cholestasis induced by chronic ethynylestradiol injections in rats. Pharmacol Biochem Behav. 2006;85:39-43. 【4.3-34】
23) Umeuchi H, Kawashima Y, Aoki CA, Kurokawa T, Nakao K, Ito M, et al. Spontaneous scratching behavior in MRL/lpr mice, a possible model for pruritus in autoimmune diseases, and antipruritic activity of a novel κ-opioid receptor agonist nalfurafine hydrochloride. Eur J Pharmacol. 2005;518:133-9. 【4.3-33】
24) Darsow U, Scharein E, Bromm B, Ring J. Skin testing of the pruritogenic activity of histamine and cytokines (interleukin-2 and tumour necrosis factor-α) at the dermal-epidermal junction. Br J Dermatol. 1997;137:415-7. 【4.3-6】
25) Kimmel M, Alscher DM, Dunst R, Braun N, Machleidt C, Kiefer T, et al. The role of micro-inflammation in the pathogenesis of uraemic pruritus in haemodialysis patients. Nephrol Dial Transplant. 2006;21:749-55. 【4.3-7】
26) Andoh T, Kuraishi Y. Nitric oxide enhances substance P-induced itch-associated responses in mice. Br J Pharmacol. 2003;138:202-8. 【4.3-8】
27) Woodward DF, Nieves AL, Friedlaender MH. Characterization of receptor subtypes involved in prostanoid-induced conjunctival pruritus and their role in mediating allergic conjunctival itching. J Pharmacol Exp Ther. 1996;279:137-42. 【4.3-9】
- 84 -

ナルフラフィン塩酸塩 2.6.2 薬理試験の概要文 Page 77
28) Pan ZZ. μ-Opposing actions of the κ-opioid receptor. Trends Pharmacol Sci. 1998;19:94-8. 【4.3-22】
29) Wakasa Y, Fujiwara A, Umeuchi H, Endoh T, Okano K, Tanaka T, et al. Inhibitory effects of TRK-820 on systemic skin scratching induced by morphine in rhesus monkeys. Life Sci. 2004;75:2947-57. 【4.3-52】
30) Endoh T, Tajima A, Suzuki T, Kamei J, Suzuki T, Narita M, et al. Characterization of the antinociceptive effects of TRK-820 in the rat. Eur J Pharmacol. 2000;387:133-40. 【4.3-57】
31) Shen S, Ingenito AJ. κ-Opioid receptors behind the blood-brain barrier are involved in the anti-hypertensive effects of systemically administered κ-agonists in the conscious spontaneously hypertensive rat. J Pharm Pharmacol. 1999;51:1251-6. 【4.3-49】
32) Wright RC, McConnaughey MM, Phan TA, Ingenito AJ. κ-Opioid receptor antisense oligonucleotide injected into rat hippocampus causes hypertension. Eur J Pharmacol. 1999;377:57-61. 【4.3-50】
33) Rao SP, Conley A, Dunbar JC. Cardiovascular responses to central administration of mu and kappa opioid receptor agonist and antagonist in normal rats. Peptides. 2003;24:745-54. 【4.3-51】
- 85 -

ナルフラフィン塩酸塩
医薬品製造販売承認申請書添付資料
2.6.3 薬理試験概要表
東レ・メディカル株式会社
- 86 -

ナルフラフィン塩酸塩 2.6.3 薬理試験概要表 Page 2
目次
2.6.3.1 薬理試験: 一覧表 ......................................................................................................... 3 2.6.3.2 効力を裏付ける試験 ..................................................................................................... 8 2.6.3.3 副次的薬理試験 ........................................................................................................... 30 2.6.3.4 安全性薬理試験 ........................................................................................................... 31 2.6.3.5 薬力学的薬物相互作用試験 ....................................................................................... 41
- 87 -

ナルフラフィン塩酸塩 2.6.3 薬理試験概要表 Page 3
2.6.3.1 薬理試験: 一覧表
被験物質: TRK-820
試験の種類 試験系 投与方法 実施施設 試験番号 記載箇所 Section
(1) 効力を裏付ける試験 1) 受容体選択性
a) げっ歯類オピオイド受容体選択性 ICR 系雄性マウス摘出輸
精管 in vitro 東レ株式会社 4.2.1.1-1
Hartley 系雄性モルモッ
ト摘出回腸 in vitro 東レ株式会社 4.2.1.1-2
b) ヒトオピオイド受容体結合性 ヒトオピオイド κ、μ、δ受容体発現細胞(HEK-293, CHO-K1, CHO)
in vitro 4.2.1.1-3
c) ヒトオピオイド受容体作動性 ヒトオピオイド κ、μ、δ受容体発現細胞(CHO-K1, CHO-dhfr(-), CHO-dhfr(-))
in vitro 東レ株式会社 4.2.1.1-4
2) 引っ掻き行動抑制作用 a) ヒスタミン誘発引っ掻き行動に対する作用
被験物質: クロルフェニラミン(対照薬) ICR 系雄性マウス 経口 東レ株式会社 4.2.1.1-5 被験物質: ケトチフェン(対照薬) ICR 系雄性マウス 経口 東レ株式会社 4.2.1.1-6 被験物質: TRK-820 ICR 系雄性マウス 経口 東レ株式会社 4.2.1.1-7
b) サブスタンス P 誘発引っ掻き行動に対する作
用
被験物質: TRK-820 ICR 系雄性マウス 経口 東レ株式会社 4.2.1.1-8 被験物質: オキサトミド(対照薬) ICR 系雄性マウス 経口 東レ株式会社 4.2.1.1-9 被験物質: クロルフェニラミン(対照薬) ICR 系雄性マウス 経口 東レ株式会社 4.2.1.1-10 被験物質: ケトチフェン(対照薬) ICR 系雄性マウス 経口 東レ株式会社 4.2.1.1-11
c) デオキシコール酸誘発引っ掻き行動に対する
作用 ICR 系雄性マウス 経口 東レ株式会社 4.2.1.1-12
- 88 -

ナルフラフィン塩酸塩 2.6.3 薬理試験概要表 Page 4
2.6.3.1 薬理試験: 一覧表(続き) 被験物質: TRK-820
試験の種類 試験系 投与方法 実施施設 試験番号 記載箇所 Section
(1) 効力を裏付ける試験 2) 引っ掻き行動抑制作用
d) モルヒネ大槽内投与誘発引っ掻き行動に対す
る作用
被験物質: TRK-820 ddY 系雄性マウス 皮下 東レ株式会社 4.2.1.1-13 被験物質: ケトチフェン(対照薬) ddY 系雄性マウス 腹腔内 東レ株式会社 4.2.1.1-14
e) 自然発症アトピー性皮膚炎モデルの引っ掻き
行動に対する作用 NC/Nga 系雄性マウス 経口 東レ株式会社 4.2.1.1-15
3) 引っ掻き行動抑制作用の持続時間 ddY 系雄性マウス 経口 東レ株式会社 4.2.1.1-16 4) 引っ掻き行動抑制作用における耐性形成能 ICR 系雄性マウス 経口 東レ株式会社 4.2.1.1-17 5) 作用機序
a) 炎症性メディエーター遊離もしくは分泌およ
び NOS 活性に対する作用 各種組織または細胞 in vitro 4.2.1.1-18
b) オピオイド受容体以外の受容体、イオンチャ
ネルまたはトランスポーターに対する作用 各種組織または細胞 in vitro 4.2.1.1-18
各種組織または細胞 in vitro 4.2.1.1-19 各種組織または細胞 in vitro 4.2.1.1-20 各種組織または細胞 in vitro 4.2.1.1-21
c) 引っ掻き行動抑制に対するオピオイド κ 受容
体拮抗薬の皮下投与の影響 ICR 系雄性マウス 経口 東レ株式会社 4.2.1.1-22
d) 引っ掻き行動抑制に対するオピオイド κ 受容
体拮抗薬の脳室内投与の影響 ddY 系雄性マウス 皮下 東レ株式会社 4.2.1.1-23
e) 局所麻酔作用の評価 Hartley 系雄性モルモッ
ト 皮内 東レ株式会社 4.2.1.1-24
- 89 -

ナルフラフィン塩酸塩 2.6.3 薬理試験概要表 Page 5
2.6.3.1 薬理試験: 一覧表(続き) 被験物質: TRK-820、10α-OH、de-CPM、de-CPM·T、NFA-G、de-CPM-G
試験の種類 試験系 投与方法 実施施設 試験番号 記載箇所 Section
(1) 効力を裏付ける試験 5) 作用機序
f) 経口剤(軟カプセル剤)中の不純物のヒト
オピオイド受容体結合性 ヒトオピオイド κ、μ、δ受容体発現細胞(HEK-293, CHO, CHO)
in vitro 4.2.1.1-25
g) 代謝物のヒトオピオイド受容体結合性 ヒトオピオイド κ、μ、δ受容体発現細胞(HEK-293, CHO-K1, CHO)
in vitro 4.2.1.1-26
h) 経口剤(軟カプセル剤)中の不純物および
代謝物のヒトオピオイド受容体作動性 ヒトオピオイド κ、μ、δ受容体発現細胞(CHO-K1, CHO-dhfr(-), CHO-dhfr(-))
in vitro 東レ株式会社 4.2.1.1-27
i) 代謝物のオピオイド受容体以外の受容体、
イオンチャネルまたはトランスポーターに
対する作用
各種組織または細胞 in vitro 4.2.1.1-28
j) 代謝物のサブスタンス P 誘発引っ掻き行動
に対する作用
被験物質: TRK-820 ddY 系雄性マウス 皮下 東レ株式会社 4.2.1.1-29 被験物質: de-CPM·T ddY 系雄性マウス 皮下 東レ株式会社 4.2.1.1-30 被験物質: NFA-G ddY 系雄性マウス 皮下 東レ株式会社 4.2.1.1-31 被験物質: de-CPM-G ddY 系雄性マウス 皮下 東レ株式会社 4.2.1.1-32
(2) 副次的薬理試験 該当なし
- 90 -

ナルフラフィン塩酸塩 2.6.3 薬理試験概要表 Page 6
2.6.3.1 薬理試験: 一覧表(続き) 被験物質: TRK-820
試験の種類 試験系 投与方法 実施施設 試験番号 記載箇所 Section
(3) 安全性薬理試験 1) 安全性薬理コアバッテリー試験
a) 中枢神経系におよぼす影響 ラットの一般症状および行動におよぼす影
響 Wistar 系雄性ラット 経口 4.2.1.3-1
b) 心血管系におよぼす影響 hERG 電流に対する作用 a hERG 導入 HEK-293 細
胞(ホールセルパッチク
ランプ法)
in vitro 4.2.1.3-2
覚醒イヌを用いた試験 a 雄性ビーグル犬 経口 4.2.1.3-3
c) 呼吸系におよぼす影響 覚醒イヌを用いた試験 a 雄性ビーグル犬 経口 4.2.1.3-3
2) 安全性薬理コアバッテリーに対するフォロー
アップ試験
a) 中枢神経系におよぼす影響 サルの一般症状および行動におよぼす影響 a 雌雄アカゲザル 静脈内 4.2.3.7.4-5
自発運動に対する作用 移所運動 ICR 系雄性マウス 経口 東レ株式会社 4.2.1.3-5 Wistar 系雄性ラット 経口 4.2.1.3-1
自発運動に対する作用 回転かご ICR 系雄性マウス 経口 東レ株式会社 4.2.1.3-6 ddY 系雄性マウス 皮下 東レ株式会社 4.2.1.3-7 協調運動に対する作用 ddY 系雄性マウス 経口 東レ株式会社 4.2.1.3-8 麻酔作用 a ICR 系雄性マウス 経口 4.2.1.3-9
a: GLP に適合した報告書
- 91 -
ナルフラフィン塩酸塩 2.6.3 薬理試験概要表 Page 7
2.6.3.1 薬理試験: 一覧表(続き) 被験物質: TRK-820
試験の種類 試験系 投与方法 実施施設 試験番号 記載箇所 Section
(3) 安全性薬理試験 2) 安全性薬理コアバッテリーに対するフォロー
アップ試験
a) 中枢神経系におよぼす影響 脳波に対する作用 a Wistar 系雄性ラット 皮下 4.2.1.3-10
鎮痛作用 ddY 系雄性マウス 経口 東レ株式会社 4.2.1.3-11 皮下 痙攣誘発および抗痙攣作用 a ICR 系雄性マウス 経口 4.2.1.3-12
正常体温に対する作用 Wistar 系雄性ラット 経口 4.2.1.3-1
b) 心血管系におよぼす影響 摘出乳頭筋の活動電位に対する作用 a Hartley 系雄性モルモッ
ト摘出乳頭筋 in vitro 4.2.1.3-13
麻酔イヌを用いた試験 a 雄性ビーグル犬 静脈内 4.2.1.3-14
3) 補足的安全性薬理試験 a) 腎/泌尿器系におよぼす影響 a SD 系雄性ラット 経口 4.2.1.3-15
b) 自律神経系におよぼす影響 a Hartley 系雄性モルモッ
ト摘出回腸 in vitro 4.2.1.3-16
c) 胃腸管系におよぼす影響 ICR 系雄性マウス 経口 東レ株式会社 4.2.1.3-17 (4) 薬力学薬物相互作用試験
1) 抗ヒスタミン薬ケトチフェンとの相互作用 ICR 系雄性マウス 皮下 東レ株式会社 4.2.1.4-1 2) 睡眠導入薬ニトラゼパムとの相互作用 ICR 系雄性マウス 皮下 東レ株式会社 4.2.1.4-2 a: GLP に適合した報告書
- 92 -
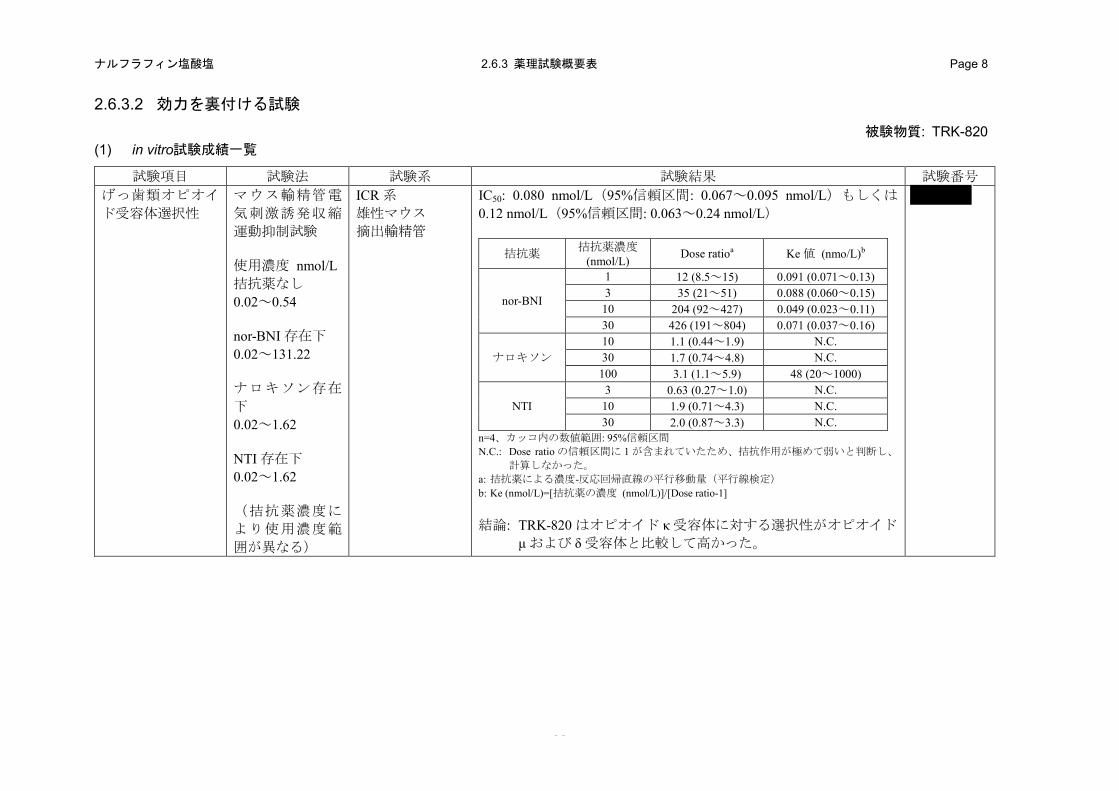
ナルフラフィン塩酸塩 2.6.3 薬理試験概要表 Page 8
2.6.3.2 効力を裏付ける試験
被験物質: TRK-820 (1) in vitro試験成績一覧
試験項目 試験法 試験系 試験結果 試験番号 げっ歯類オピオイ
ド受容体選択性 マウス輸精管電
気刺激誘発収縮
運動抑制試験 使用濃度 nmol/L 拮抗薬なし 0.02~0.54 nor-BNI 存在下 0.02~131.22 ナロキソン存在
下 0.02~1.62 NTI 存在下 0.02~1.62 (拮抗薬濃度に
より使用濃度範
囲が異なる)
ICR 系 雄性マウス 摘出輸精管
IC50: 0.080 nmol/L(95%信頼区間: 0.067~0.095 nmol/L)もしくは
0.12 nmol/L(95%信頼区間: 0.063~0.24 nmol/L)
拮抗薬 拮抗薬濃度 (nmol/L) Dose ratioa Ke 値 (nmo/L)b
nor-BNI
1 12 (8.5~15) 0.091 (0.071~0.13) 3 35 (21~51) 0.088 (0.060~0.15)
10 204 (92~427) 0.049 (0.023~0.11) 30 426 (191~804) 0.071 (0.037~0.16)
ナロキソン 10 1.1 (0.44~1.9) N.C. 30 1.7 (0.74~4.8) N.C.
100 3.1 (1.1~5.9) 48 (20~1000)
NTI 3 0.63 (0.27~1.0) N.C.
10 1.9 (0.71~4.3) N.C. 30 2.0 (0.87~3.3) N.C.
n=4、カッコ内の数値範囲: 95%信頼区間 N.C.: Dose ratio の信頼区間に 1 が含まれていたため、拮抗作用が極めて弱いと判断し、
計算しなかった。 a: 拮抗薬による濃度-反応回帰直線の平行移動量(平行線検定) b: Ke (nmol/L)=[拮抗薬の濃度 (nmol/L)]/[Dose ratio-1] 結論: TRK-820 はオピオイド κ 受容体に対する選択性がオピオイド
μ および δ 受容体と比較して高かった。
- 93 -

ナルフラフィン塩酸塩 2.6.3 薬理試験概要表 Page 9
2.6.3.2 効力を裏付ける試験
被験物質: TRK-820 (1) in vitro 試験成績一覧(続き)
試験項目 試験法 試験系 試験結果 試験番号 げっ歯類オピオイ
ド受容体選択性
(続き)
モルモット回腸
電気刺激誘発収
縮運動抑制試験 使用濃度 nmol/L 拮抗薬なし 0.0003~0.0243 nor-BNI 存在下 0.01~16.2 ナロキソン存在
下 0.001~0.162 (拮抗薬濃度に
より使用濃度範
囲が異なる)
Hartley 系 雄性モルモット 摘出回腸
IC50: 0.0081 nmol/L(95%信頼区間: 0.0057~0.011 nmol/L)
拮抗薬 拮抗薬濃度 (nmol/L) Dose ratioa Ke 値 (nmo/L)b
nor-BNI
3 21 (13~34)
0.15 (0.090~0.24)
10 143 (94~225)
0.070 (0.045~0.11)
30 347 (217~593)
0.087 (0.051~0.14)
ナロキソン
10 3.9 (2.0~8.0)
3.4 (1.4~9.6)
30 6.1 (3.9~10)
5.8 (3.2~10)
100 19 (11~38)
5.5 (2.7~9.6)
n=4、カッコ内の数値範囲: 95%信頼区間 a: 拮抗薬による濃度-反応回帰直線の平行移動量(平行線検定) b: Ke (nmol/L)=[拮抗薬の濃度 (nmol/L)]/[Dose ratio-1] 結論: TRK-820 はオピオイド κ 受容体に対する選択性がオピオイド
μ 受容体と比較して高かった。
- 94 -

ナルフラフィン塩酸塩 2.6.3 薬理試験概要表 Page 10
2.6.3.2 効力を裏付ける試験
被験物質: TRK-820 (1) in vitro 試験成績一覧(続き)
試験項目 試験法 試験系 試験結果 試験番号 ヒトオピオイド受
容体結合性 受容体結合試験 使用濃度 κ: 0.03~10 μ: 0.3~300 δ: 30~30000 nmol/L
ヒトオピオイド受
容体強制発現細胞 κ: HEK-293 μ: CHO-K1 δ: CHO
Ki 値 (nmol/L)a
オピオイド κ 受容体 オピオイド μ 受容体 オピオイド δ 受容体 0.244 ± 0.0256 2.21 ± 0.214 484 ± 59.6
a: 3 回の実験の平均値 ± 標準誤差 結論: TRK-820 はオピオイド μ および δ 受容体と比較して、それ
ぞれ 9 および 1980 倍、オピオイド κ 受容体に対する結合
性が強かった。
- 95 -
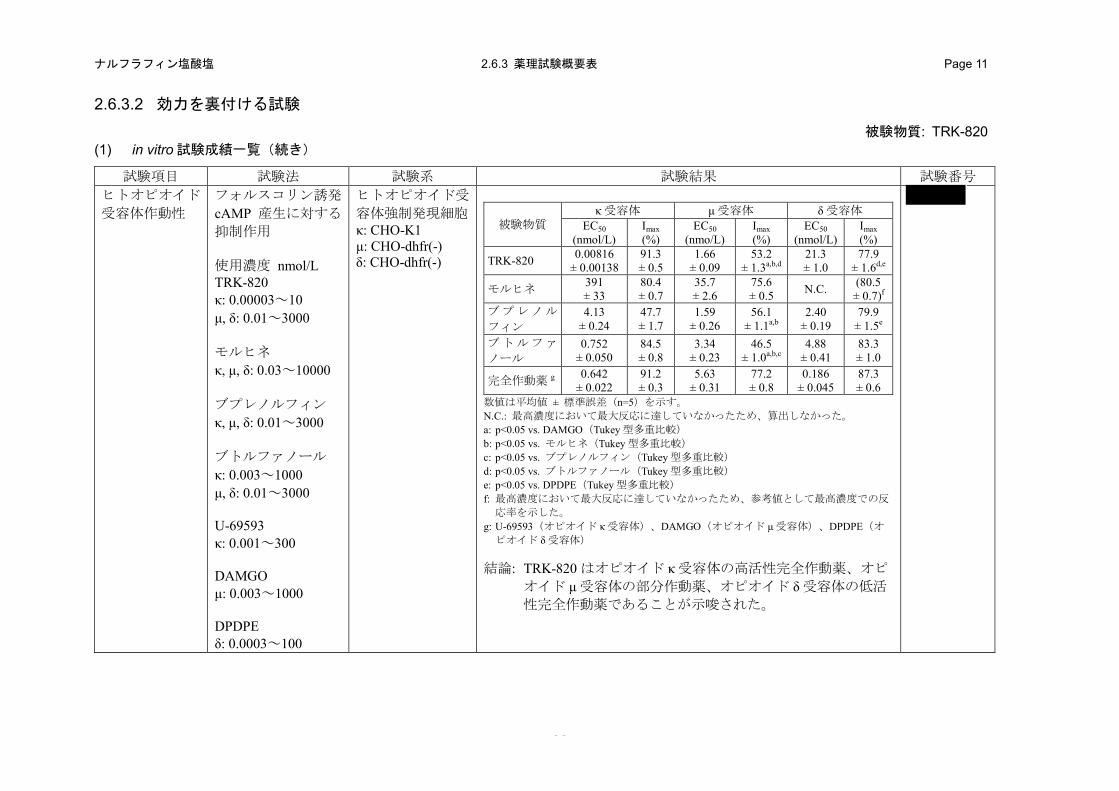
ナルフラフィン塩酸塩 2.6.3 薬理試験概要表 Page 11
2.6.3.2 効力を裏付ける試験
被験物質: TRK-820 (1) in vitro 試験成績一覧(続き)
試験項目 試験法 試験系 試験結果 試験番号 ヒトオピオイド
受容体作動性 フォルスコリン誘発
cAMP 産生に対する
抑制作用 使用濃度 nmol/L TRK-820 κ: 0.00003~10 μ, δ: 0.01~3000 モルヒネ κ, μ, δ: 0.03~10000 ブプレノルフィン κ, μ, δ: 0.01~3000 ブトルファノール κ: 0.003~1000 μ, δ: 0.01~3000 U-69593 κ: 0.001~300 DAMGO μ: 0.003~1000 DPDPE δ: 0.0003~100
ヒトオピオイド受
容体強制発現細胞 κ: CHO-K1 μ: CHO-dhfr(-) δ: CHO-dhfr(-)
被験物質 κ 受容体 μ 受容体 δ 受容体
EC50 (nmol/L)
Imax (%)
EC50 (nmo/L)
Imax (%)
EC50 (nmol/L)
Imax (%)
TRK-820 0.00816 ± 0.00138
91.3 ± 0.5
1.66 ± 0.09
53.2 ± 1.3a,b,d
21.3 ± 1.0
77.9 ± 1.6d,e
モルヒネ 391 ± 33
80.4 ± 0.7
35.7 ± 2.6
75.6 ± 0.5 N.C. (80.5
± 0.7)f ブプレノル
フィン 4.13
± 0.24 47.7 ± 1.7
1.59 ± 0.26
56.1 ± 1.1a,b
2.40 ± 0.19
79.9 ± 1.5e
ブトルファ
ノール 0.752
± 0.050 84.5 ± 0.8
3.34 ± 0.23
46.5 ± 1.0a,b,c
4.88 ± 0.41
83.3 ± 1.0
完全作動薬 g 0.642 ± 0.022
91.2 ± 0.3
5.63 ± 0.31
77.2 ± 0.8
0.186 ± 0.045
87.3 ± 0.6
数値は平均値 ± 標準誤差(n=5)を示す。 N.C.: 最高濃度において最大反応に達していなかったため、算出しなかった。 a: p<0.05 vs. DAMGO(Tukey 型多重比較) b: p<0.05 vs. モルヒネ(Tukey 型多重比較) c: p<0.05 vs. ブプレノルフィン(Tukey 型多重比較) d: p<0.05 vs. ブトルファノール(Tukey 型多重比較) e: p<0.05 vs. DPDPE(Tukey 型多重比較) f: 最高濃度において最大反応に達していなかったため、参考値として最高濃度での反
応率を示した。 g: U-69593(オピオイド κ 受容体)、DAMGO(オピオイド μ 受容体)、DPDPE(オ
ピオイド δ 受容体) 結論: TRK-820 はオピオイド κ 受容体の高活性完全作動薬、オピ
オイド μ 受容体の部分作動薬、オピオイド δ 受容体の低活
性完全作動薬であることが示唆された。
- 96 -

ナルフラフィン塩酸塩 2.6.3 薬理試験概要表 Page 12
2.6.3.2 効力を裏付ける試験
被験物質: TRK-820 (2) in vivo試験成績一覧
試験項目 試験目的 試験系 投与 経路 試験結果 試験番号
ヒスタミン誘発
引っ掻き行動に
対する作用
抗ヒスタミン薬が
有効な痒みに対す
る止痒作用を検討
ICR 系 雄性マウス
経口 ED50 (95%信頼区間)
TRK-820 用量: 3, 10, 30, 100 μg/kg
ケトチフェン 用量: 0.3, 3, 30 mg/kg
クロルフェニラミン 用量: 3, 10, 30 mg/kg
7.30 μg/kg (4.22~12.6 μg/kg)
3.35 mg/kg (0.554~20.3 mg/kg)
8.50 mg/kg (1.73~25.5 mg/kg)
起痒剤のヒスタミンは 10 μg/50 μL/site で背部皮内に投与した。 結論: TRK-820 は抗ヒスタミン薬が有効な痒みに対して止痒作
用を有する可能性が示唆された。 サブスタンス P誘発引っ掻き行
動に対する作用
抗ヒスタミン薬が
効き難い痒みに対
する止痒作用を検
討
ICR 系 雄性マウス
経口 ED50 (95%信頼区間)
TRK-820 用量: 3, 10, 30, 100 μg/kg
ケトチフェン 用量: 0.1, 1, 10, 100
mg/kg
クロルフェ
ニラミン 用量: 1, 3, 10,
30 mg/kg
オキサトミ
ド 用量: 0.1, 1,
10, 100 mg/kg 19.6 μg/kg
(9.59~40.0 μg/kg) 9.61 mg/kg
(0.541~171 mg/kg) (ただし、100 mg/kg でも統計学的に有
意な作用なし)
作用なし 作用なし
起痒剤のサブスタンス P は 250 nmol/50 μL/site で背部皮内に投与した。 結論: TRK-820 は抗ヒスタミン薬が十分な効果を示さない痒み
に対して止痒作用を有する可能性が示唆された。
- 97 -

ナルフラフィン塩酸塩 2.6.3 薬理試験概要表 Page 13
2.6.3.2 効力を裏付ける試験
被験物質: TRK-820 (2) in vivo 試験成績一覧(続き)
試験項目 試験目的 試験系 投与 経路 試験結果 試験番号
デオキシコール
酸誘発引っ掻き
行動に対する作
用
胆汁うっ滞性の痒
みに対する止痒作
用を検討
ICR 系 雄性マウス
経口 ED50 (95%信頼区間)
TRK-820 用量: 3, 10, 30, 100 μg/kg
ケトチフェン 用量: 1, 3, 10, 30
mg/kg
ナルトレキソン 用量: 0.3, 1, 3, 10 mg/kg
7.62 μg/kg (3.91~12.0 μg/kg) 作用なし
0.173 mg/kg (引っ掻き行動抑制率は
60~70%で頭打ち) 起痒剤のデオキシコール酸は 100 μg/50 μL/site で背部皮内に投与した。 ナルトレキソンは皮下投与した。 結論: TRK-820 は抗ヒスタミン薬が無効な胆汁うっ滞性の痒み
に対して止痒作用を有する可能性が示唆された。 モルヒネ大槽内
投与誘発引っ掻
き行動に対する
作用
中枢神経系のオピ
オイド μ 受容体の
活性化に伴う痒み
に対する止痒作用
を検討
ddY 系 雄性マウス
皮下 ED50 (95%信頼区間)
TRK-820 用量: 1.25, 2.5, 5, 10 μg/kg
ケトチフェン 用量: 0.01, 0.1, 1, 10 mg/kg
2.34 μg/kg (1.28~3.34 μg/kg) 作用なし 起痒剤のモルヒネは 0.3 nmol/5 μL/site で大槽内に投与した。 ケトチフェンは腹腔内投与した。 結論: TRK-820 は抗ヒスタミン薬が無効な中枢神経系のオピオ
イド μ 受容体の活性化に伴う痒みに対して止痒作用を有
する可能性が示唆された。
- 98 -

ナルフラフィン塩酸塩 2.6.3 薬理試験概要表 Page 14
2.6.3.2 効力を裏付ける試験
被験物質: TRK-820 (2) in vivo 試験成績一覧(続き)
試験項目 試験目的 試験系 投与 経路 試験結果 試験番号
自然発症アトピ
ー性皮膚炎モデ
ルの引っ掻き行
動に対する作用
アトピー性皮膚炎
に伴う痒みに対す
る止痒作用を検討
NC/Nga 系 雄性マウス
経口 ED50 (95%信頼区間)
TRK-820 用量: 10, 30, 100 μg/kg 46.1 μg/kg (25.7~125 μg/kg)
結論: TRK-820 はアトピー性皮膚炎に伴う痒みに対して止痒作
用を有する可能性が示唆された。なお、本モデルの引っ
掻き行動は抗ヒスタミン薬のクロルフェニラミンもしく
はケトチフェンで抑制できないことが報告されている。 引っ掻き行動抑
制作用の持続時
間
経口投与による止
痒作用の持続時間
を検討
ddY 系 雄性マウス
経口 TRK-820(100 μg/kg)経口投与の 0.5、2、4 および 6 時間後で
は統計学的に有意にサブスタンス P(250 nmol/50 μL/site)皮内
投与誘発引っ掻き行動が抑制された。一方、TRK-820 経口投与
の 8 時間後では統計学的に有意な差は消失した。 結論: マウスに TRK-820 を経口投与したときの止痒作用の持続
時間は約 6 時間であることが示唆された。
- 99 -
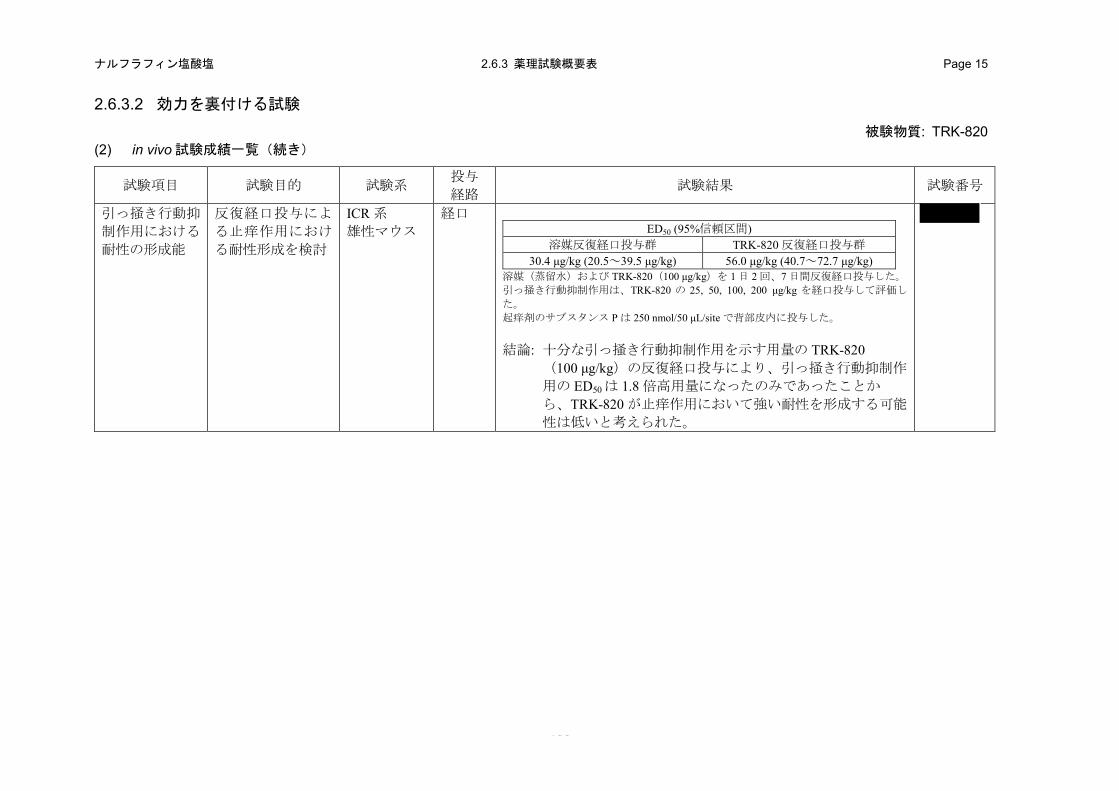
ナルフラフィン塩酸塩 2.6.3 薬理試験概要表 Page 15
2.6.3.2 効力を裏付ける試験
被験物質: TRK-820 (2) in vivo 試験成績一覧(続き)
試験項目 試験目的 試験系 投与 経路 試験結果 試験番号
引っ掻き行動抑
制作用における
耐性の形成能
反復経口投与によ
る止痒作用におけ
る耐性形成を検討
ICR 系 雄性マウス
経口 ED50 (95%信頼区間)
溶媒反復経口投与群 TRK-820 反復経口投与群 30.4 μg/kg (20.5~39.5 μg/kg) 56.0 μg/kg (40.7~72.7 μg/kg)
溶媒(蒸留水)および TRK-820(100 μg/kg)を 1 日 2 回、7 日間反復経口投与した。 引っ掻き行動抑制作用は、TRK-820 の 25, 50, 100, 200 μg/kg を経口投与して評価し
た。 起痒剤のサブスタンス P は 250 nmol/50 μL/site で背部皮内に投与した。 結論: 十分な引っ掻き行動抑制作用を示す用量の TRK-820
(100 μg/kg)の反復経口投与により、引っ掻き行動抑制作
用の ED50 は 1.8 倍高用量になったのみであったことか
ら、TRK-820 が止痒作用において強い耐性を形成する可能
性は低いと考えられた。
- 100 -
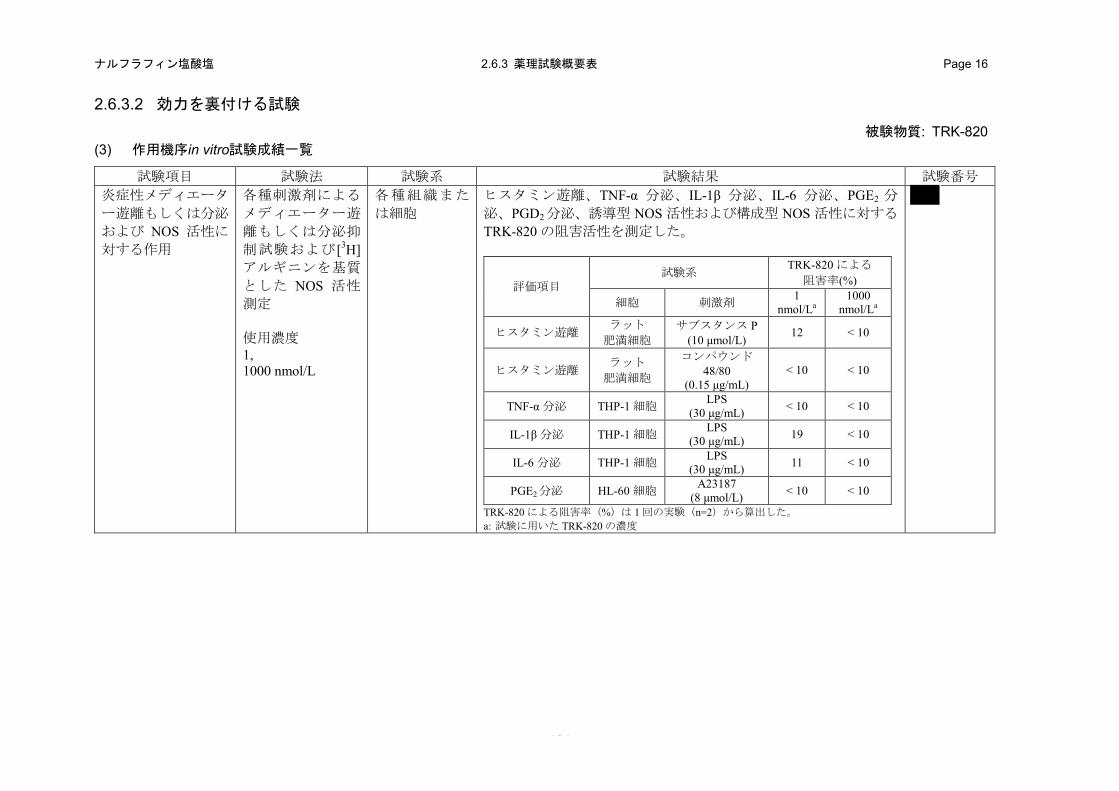
ナルフラフィン塩酸塩 2.6.3 薬理試験概要表 Page 16
2.6.3.2 効力を裏付ける試験
被験物質: TRK-820 (3) 作用機序in vitro試験成績一覧
試験項目 試験法 試験系 試験結果 試験番号 炎症性メディエータ
ー遊離もしくは分泌
および NOS 活性に
対する作用
各種刺激剤による
メディエーター遊
離もしくは分泌抑
制試験および [3H]アルギニンを基質
とした NOS 活性
測定 使用濃度 1, 1000 nmol/L
各種組織また
は細胞 ヒスタミン遊離、TNF-α 分泌、IL-1β 分泌、IL-6 分泌、PGE2 分
泌、PGD2分泌、誘導型 NOS 活性および構成型 NOS 活性に対する
TRK-820 の阻害活性を測定した。
評価項目 試験系 TRK-820 による
阻害率(%)
細胞 刺激剤 1 nmol/La
1000 nmol/La
ヒスタミン遊離 ラット 肥満細胞
サブスタンス P (10 μmol/L) 12 < 10
ヒスタミン遊離 ラット 肥満細胞
コンパウンド48/80
(0.15 μg/mL) < 10 < 10
TNF-α 分泌 THP-1 細胞 LPS (30 μg/mL) < 10 < 10
IL-1β 分泌 THP-1 細胞 LPS (30 μg/mL) 19 < 10
IL-6 分泌 THP-1 細胞 LPS (30 μg/mL) 11 < 10
PGE2分泌 HL-60 細胞 A23187 (8 μmol/L) < 10 < 10
TRK-820 による阻害率(%)は 1 回の実験(n=2)から算出した。 a: 試験に用いた TRK-820 の濃度
- 101 -

ナルフラフィン塩酸塩 2.6.3 薬理試験概要表 Page 17
2.6.3.2 効力を裏付ける試験
被験物質: TRK-820 (3) 作用機序 in vitro 試験成績一覧(続き)
試験項目 試験法 試験系 試験結果 試験番号 炎症性メディエータ
ー遊離もしくは分泌
および NOS 活性に
対する作用(続き)
各種刺激剤による
メディエーター遊
離もしくは分泌抑
制試験および [3H]アルギニンを基質
とした NOS 活性
測定 使用濃度 1, 1000 nmol/L
各種組織また
は細胞
評価項目 試験系 TRK-820 による
阻害率(%)
細胞 刺激剤 1 nmol/La
1000 nmol/La
PGD2 分泌 ラット肥満
細胞 A23187
(10 μmol/L) < 10 < 10
誘導型 NOS 活性 RAW264-7
細胞 [3H]アルギニン
(28 nmol/L) N.E. < 10
構成型 NOS 活性 HUVEC 細胞
[3H]アルギニン(56 nmol/L) N.E. < 10
TRK-820 による阻害率(%)は 1 回の実験(n=2)から算出した。 N.E.: 実施せず a: 試験に用いた TRK-820 の濃度 結論: TRK-820 はすべての炎症性メディエーター遊離ならびに誘
導型および構成型 NOS 活性に対して阻害作用を示さなかっ
たことから、TRK-820 の止痒作用は、炎症性メディエータ
ー遊離および NOS 活性に対する阻害作用には起因しないこ
とが示唆された。
- 102 -

ナルフラフィン塩酸塩 2.6.3 薬理試験概要表 Page 18
2.6.3.2 効力を裏付ける試験
被験物質: TRK-820 (3) 作用機序 in vitro 試験成績一覧(続き)
試験項目 試験法 試験系 試験結果 試験番号 オピオイド受容体以
外の受容体、イオン
チャネルまたはトラ
ンスポーターに対す
る作用
各受容体、イオン
チャネルおよびト
ランスポーターへ
の結合試験 使用濃度 1000, 10000 nmol/L
各種組織また
は細胞
受容体 イオンチャネル
トランスポーター
阻害率 (%) TRK-820
1000 nmol/L TRK-820
10000 nmol/L Adenosine A1 N.E. < 10 Adenosine A2A N.E. 13 Transporter, Adenosine 4 N.E. Adrenergic α1, Non-Selective N.E. 27 Adrenergic α2, Non-Selective N.E. 11 Adrenergic β1 -4 N.E. Adrenergic β2 -3 N.E. Transporter, Norepinephrine (NET) -5 N.E. Angiotensin II N.E. < 10 Atrial natriuretic peptide (ANP) < 10 N.E. Bombesin N.E. 12 Bradykinin B2 4 N.E. Calcitonin gene related peptide (CGRP) 10 N.E.
Calcium channel Type L, Dihydropyridine site N.E. < 10
Calcium Channel N-Type 6 N.E. Cannabinoid CB1 -1 N.E. CC Chemokine Receptor CCR1 < 10 N.E. CC Chemokine Receptor CCR2 < 10 N.E. Cholecystokinin A N.E. < 10 Cholecystokinin B N.E. < 10 Transporter, Choline -2 N.E. Corticotropin Releasing Factor CRF1
-3 N.E.
阻害率(%)は 1 回の実験(n=2)から算出した。 N.E.: 実施せず
- 103 -

ナルフラフィン塩酸塩 2.6.3 薬理試験概要表 Page 19
2.6.3.2 効力を裏付ける試験
被験物質: TRK-820 (3) 作用機序 in vitro 試験成績一覧(続き)
試験項目 試験法 試験系 試験結果 試験番号 オピオイド受容体以
外の受容体、イオン
チャネルまたはトラ
ンスポーターに対す
る作用(続き)
各受容体、イオン
チャネルおよびト
ランスポーターへ
の結合試験 使用濃度 1000, 10000 nmol/L
各種組織およ
び細胞
受容体 イオンチャネル
トランスポーター
阻害率 (%) TRK-820
1000 nmol/L TRK-820
10000 nmol/L Dopamine D1 N.E. < 10 Dopamine D2 N.E. 20 Dopamine D3 9 N.E. Transporter, Dopamine (DAT) -11 N.E. Transporter, GABA -3 N.E. GABAA, Agonist Site N.E. < 10 GABAA, Flunitrazepam, Central -1 N.E. GABAA, Chloride Channel, TBPS 12 N.E. GABAB, Non-Selective 7 N.E. Glucocorticoid 1 N.E. Glutamate, Non-Selective N.E. < 10 Glutamate, AMPA N.E. < 10 Glutamate, Kainate N.E. < 10 Glutamate, NMDA, Agonist Site N.E. 10 Glutamate, NMDA, Phencyclidine Site N.E. < 10
Glutamate, Metabotropic, mGlu2 -1 N.E. Glutamate, Metabotropic, mGlu5 13 N.E. Glycine, Strychnine-Sensitive 5 N.E. Transporter, Glycine 0 N.E.
阻害率(%)は 1 回の実験(n=2)から算出した。 N.E.: 実施せず
- 104 -
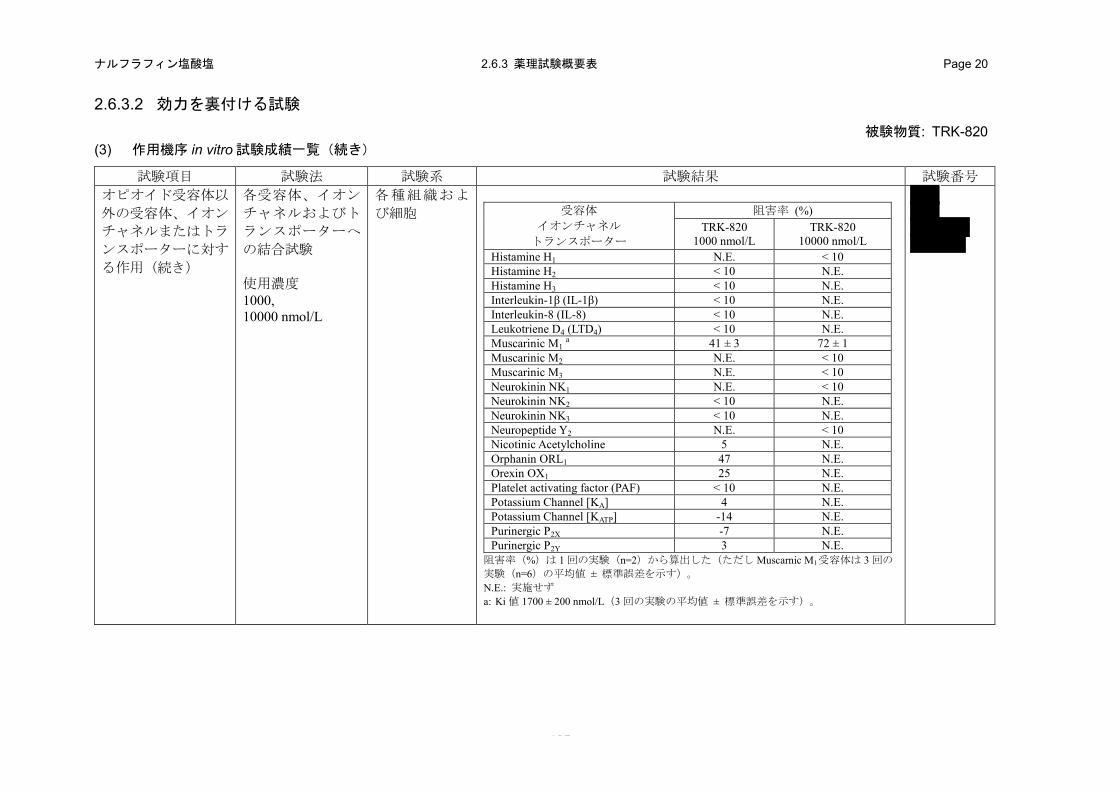
ナルフラフィン塩酸塩 2.6.3 薬理試験概要表 Page 20
2.6.3.2 効力を裏付ける試験
被験物質: TRK-820 (3) 作用機序 in vitro 試験成績一覧(続き)
試験項目 試験法 試験系 試験結果 試験番号 オピオイド受容体以
外の受容体、イオン
チャネルまたはトラ
ンスポーターに対す
る作用(続き)
各受容体、イオン
チャネルおよびト
ランスポーターへ
の結合試験 使用濃度 1000, 10000 nmol/L
各種組織およ
び細胞
受容体 イオンチャネル
トランスポーター
阻害率 (%) TRK-820
1000 nmol/L TRK-820
10000 nmol/L Histamine H1 N.E. < 10 Histamine H2 < 10 N.E. Histamine H3 < 10 N.E. Interleukin-1β (IL-1β) < 10 N.E. Interleukin-8 (IL-8) < 10 N.E. Leukotriene D4 (LTD4) < 10 N.E. Muscarinic M1 a 41 ± 3 72 ± 1 Muscarinic M2 N.E. < 10 Muscarinic M3 N.E. < 10 Neurokinin NK1 N.E. < 10 Neurokinin NK2 < 10 N.E. Neurokinin NK3 < 10 N.E. Neuropeptide Y2 N.E. < 10 Nicotinic Acetylcholine 5 N.E. Orphanin ORL1 47 N.E. Orexin OX1 25 N.E. Platelet activating factor (PAF) < 10 N.E. Potassium Channel [KA] 4 N.E. Potassium Channel [KATP] -14 N.E. Purinergic P2X -7 N.E. Purinergic P2Y 3 N.E.
阻害率(%)は 1 回の実験(n=2)から算出した(ただし Muscarnic M1受容体は 3 回の
実験(n=6)の平均値 ± 標準誤差を示す)。 N.E.: 実施せず a: Ki 値 1700 ± 200 nmol/L(3 回の実験の平均値 ± 標準誤差を示す)。
- 105 -
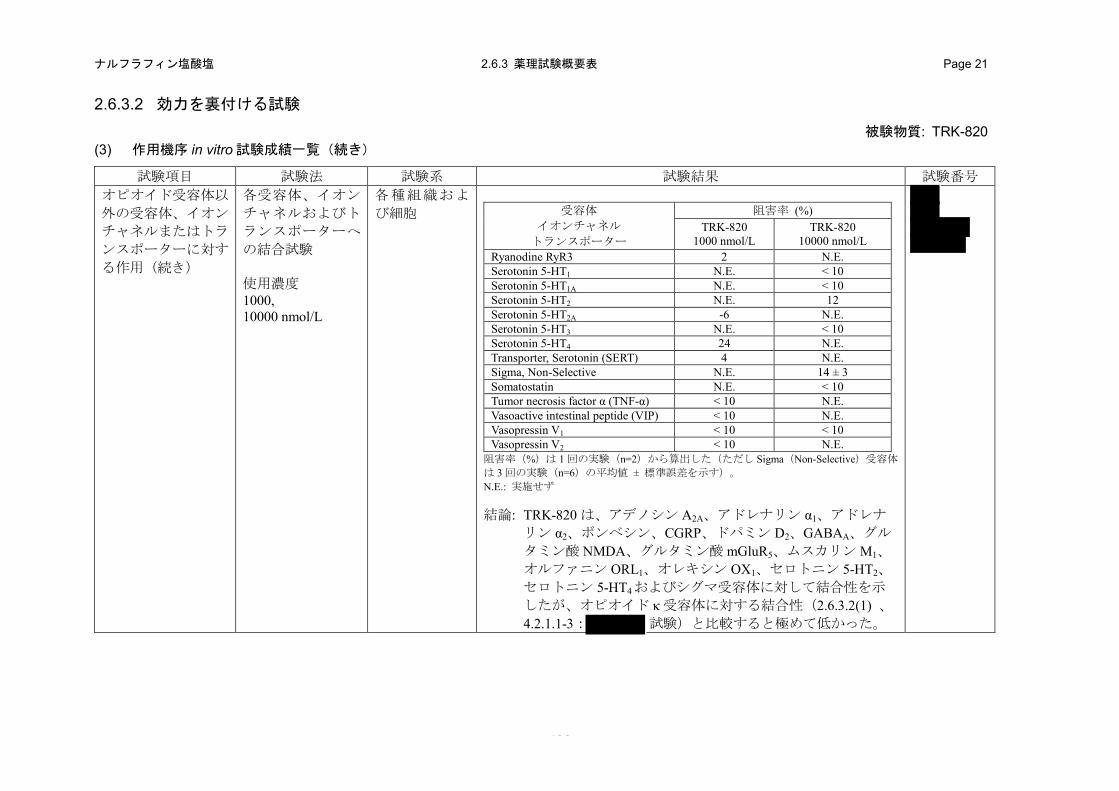
ナルフラフィン塩酸塩 2.6.3 薬理試験概要表 Page 21
2.6.3.2 効力を裏付ける試験
被験物質: TRK-820 (3) 作用機序 in vitro 試験成績一覧(続き)
試験項目 試験法 試験系 試験結果 試験番号 オピオイド受容体以
外の受容体、イオン
チャネルまたはトラ
ンスポーターに対す
る作用(続き)
各受容体、イオン
チャネルおよびト
ランスポーターへ
の結合試験 使用濃度 1000, 10000 nmol/L
各種組織およ
び細胞
受容体 イオンチャネル
トランスポーター
阻害率 (%) TRK-820
1000 nmol/L TRK-820
10000 nmol/L Ryanodine RyR3 2 N.E. Serotonin 5-HT1 N.E. < 10 Serotonin 5-HT1A N.E. < 10 Serotonin 5-HT2 N.E. 12 Serotonin 5-HT2A -6 N.E. Serotonin 5-HT3 N.E. < 10 Serotonin 5-HT4 24 N.E. Transporter, Serotonin (SERT) 4 N.E. Sigma, Non-Selective N.E. 14 ± 3 Somatostatin N.E. < 10 Tumor necrosis factor α (TNF-α) < 10 N.E. Vasoactive intestinal peptide (VIP) < 10 N.E. Vasopressin V1 < 10 < 10 Vasopressin V2 < 10 N.E.
阻害率(%)は 1 回の実験(n=2)から算出した(ただし Sigma(Non-Selective)受容体
は 3 回の実験(n=6)の平均値 ± 標準誤差を示す)。 N.E.: 実施せず
結論: TRK-820 は、アデノシン A2A、アドレナリン α1、アドレナ
リン α2、ボンベシン、CGRP、ドパミン D2、GABAA、グル
タミン酸 NMDA、グルタミン酸 mGluR5、ムスカリン M1、
オルファニン ORL1、オレキシン OX1、セロトニン 5-HT2、
セロトニン 5-HT4およびシグマ受容体に対して結合性を示
したが、オピオイド κ 受容体に対する結合性(2.6.3.2(1) 、4.2.1.1-3: 試験)と比較すると極めて低かった。
- 106 -

ナルフラフィン塩酸塩 2.6.3 薬理試験概要表 Page 22
2.6.3.2 効力を裏付ける試験
被験物質: TRK-820 (4) 作用機序in vivo試験成績一覧
試験項目 試験目的 試験系 投与 経路 試験結果 試験番号
引っ掻き行動の抑制作用
に対するオピオイド κ 受
容体拮抗薬の皮下投与の
影響
止痒作用発現におけ
るオピオイド κ 受容
体の関与を検討
ICR 系 雄性マウス
経口 TRK-820(100 μg/kg)経口投与によるサブスタンス
P(250 nmol/50 μL/site)皮内投与誘発引っ掻き行動
の抑制作用はオピオイド κ 受容体拮抗薬 nor-BNI(1、3 および 10 mg/kg)皮下投与により用量依存
的に拮抗され、10 mg/kg では TRK-820 の統計学的
に有意な引っ掻き行動抑制作用が消失した。 結論: TRK-820 はオピオイド κ 受容体の活性化を介
して止痒作用を発現することが示唆された。 引っ掻き行動の抑制作用
に対するオピオイド κ 受
容体拮抗薬の脳室内投与
の影響
止痒作用発現におけ
る中枢神経系のオピ
オイド κ 受容体の関
与を検討
ddY 系 雄性マウス
皮下 TRK-820(10 μg/kg)皮下投与によるサブスタンス P(250 nmol/50 μL/site)皮内投与誘発引っ掻き行動の
抑制作用はオピオイド κ 受容体拮抗薬 nor-BNI(10 μg/site)脳室内投与により拮抗され、TRK-820の統計学的に有意な引っ掻き行動抑制作用が消失し
た。 結論: TRK-820 は中枢神経系のオピオイド κ 受容体
の活性化を介して止痒作用を発現することが
示唆された。 局所麻酔作用の評価 局所麻酔作用の有無
を検討 Hartley 系 雄性モルモ
ット
皮内 TRK-820 の 0.01、0.1、1 および 10 μg/mL を
200 μL/site の容量で皮内投与してもマンドリン線刺
激による皮膚の攣縮反応は抑制されなかった。 結論: TRK-820 は局所麻酔作用を有しておらず、止
痒作用発現は局所麻酔作用に起因していない
ことが示唆された。
- 107 -

ナルフラフィン塩酸塩 2.6.3 薬理試験概要表 Page 23
2.6.3.2 効力を裏付ける試験
被験物質: 10α-OH (5) 作用機序in vitro試験成績一覧(経口剤中の不純物の薬理作用)
試験項目 試験法 試験系 試験結果 試験番号 経口剤(軟カプセル剤)
中の不純物のヒトオピオ
イド受容体結合性
受容体結合試験 使用濃度 κ: 1~10000 μ: 300~100000 δ: 10000 nmol/L
ヒトオピオイド受
容体強制発現細胞 κ: HEK-293 μ: CHO δ: CHO
項目 10α-OH
κ 受容体 μ 受容体 δ 受容体 結合阻害率 (%) (10000 nmol/L)a 100 66 8
Ki値 (nmol/L)b 4.26 2070 > 10000 c a: 10α-OH の 10000 nmol/L 適用時の阻害率。数値は 1 回の実験(n=2)から算
出した。 b: 数値は 1 回の実験(n=2)から算出した。 c: 10α-OH の 10000 nmol/L 適用時の阻害率が 50%未満であったため、Ki値を算
出する実験は実施していない。 結論: 経口剤中の不純物 10α-OH のオピオイド κ および μ 受
容体に対する Ki 値は TRK-820(2.6.3.2(1)、4.2.1.1-3:試験)と比較してそれぞれ 17 および 937 倍
高値であったことから、結合性が低いことが示唆され
た。また、オピオイド δ 受容体にはほとんど結合性を
示さなかった。
- 108 -
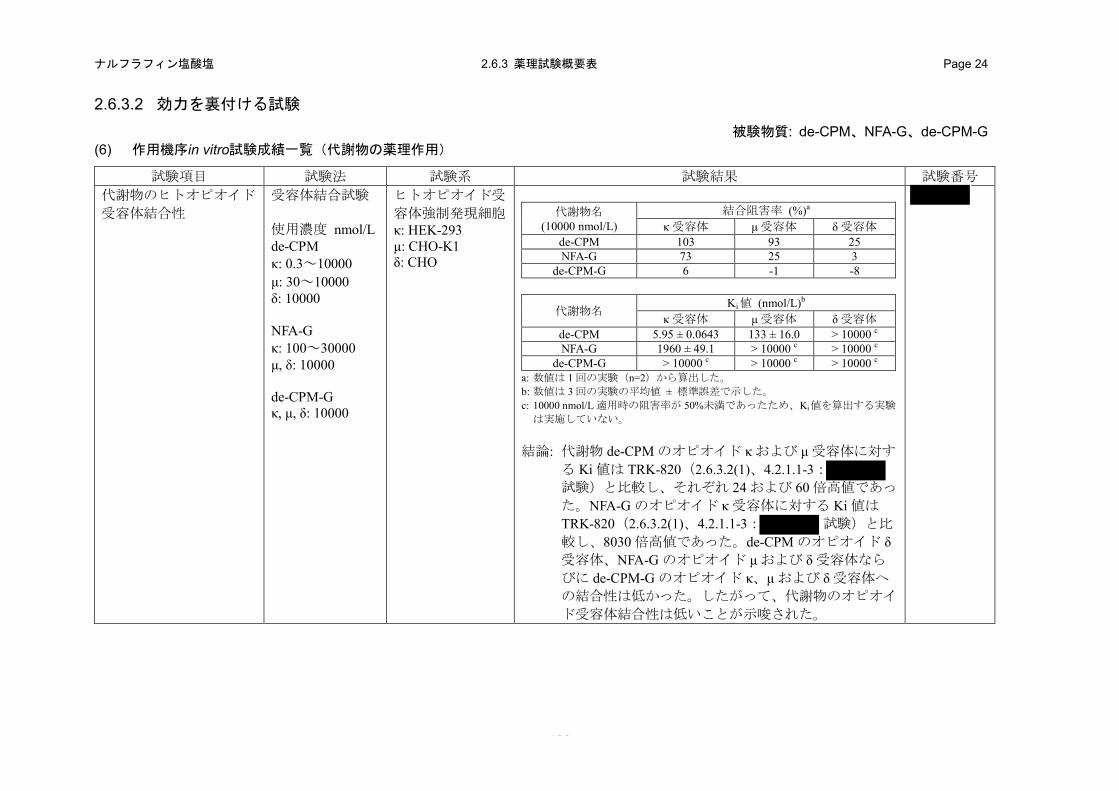
ナルフラフィン塩酸塩 2.6.3 薬理試験概要表 Page 24
2.6.3.2 効力を裏付ける試験
被験物質: de-CPM、NFA-G、de-CPM-G (6) 作用機序in vitro試験成績一覧(代謝物の薬理作用)
試験項目 試験法 試験系 試験結果 試験番号 代謝物のヒトオピオイド
受容体結合性 受容体結合試験 使用濃度 nmol/L de-CPM κ: 0.3~10000 μ: 30~10000 δ: 10000 NFA-G κ: 100~30000 μ, δ: 10000 de-CPM-G κ, μ, δ: 10000
ヒトオピオイド受
容体強制発現細胞 κ: HEK-293 μ: CHO-K1 δ: CHO
代謝物名
(10000 nmol/L) 結合阻害率 (%)a
κ 受容体 μ 受容体 δ 受容体 de-CPM 103 93 25 NFA-G 73 25 3
de-CPM-G 6 -1 -8
代謝物名 Ki値 (nmol/L)b
κ 受容体 μ 受容体 δ 受容体 de-CPM 5.95 ± 0.0643 133 ± 16.0 > 10000 c NFA-G 1960 ± 49.1 > 10000 c > 10000 c
de-CPM-G > 10000 c > 10000 c > 10000 c a: 数値は 1 回の実験(n=2)から算出した。 b: 数値は 3 回の実験の平均値 ± 標準誤差で示した。 c: 10000 nmol/L 適用時の阻害率が 50%未満であったため、Ki値を算出する実験
は実施していない。 結論: 代謝物 de-CPM のオピオイド κ および μ 受容体に対す
る Ki 値は TRK-820(2.6.3.2(1)、4.2.1.1-3:試験)と比較し、それぞれ 24 および 60 倍高値であっ
た。NFA-G のオピオイド κ 受容体に対する Ki 値は
TRK-820(2.6.3.2(1)、4.2.1.1-3: 試験)と比
較し、8030 倍高値であった。de-CPM のオピオイド δ受容体、NFA-G のオピオイド μ および δ 受容体なら
びに de-CPM-G のオピオイド κ、μ および δ 受容体へ
の結合性は低かった。したがって、代謝物のオピオイ
ド受容体結合性は低いことが示唆された。
- 109 -
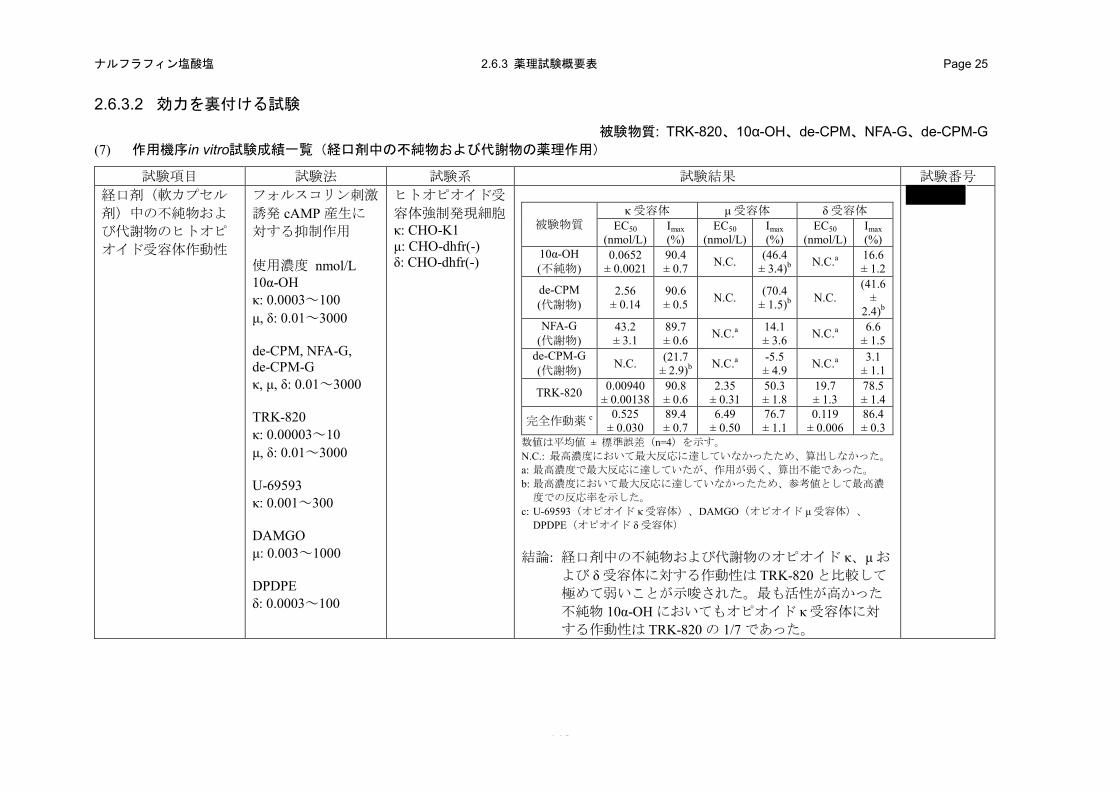
ナルフラフィン塩酸塩 2.6.3 薬理試験概要表 Page 25
2.6.3.2 効力を裏付ける試験
被験物質: TRK-820、10α-OH、de-CPM、NFA-G、de-CPM-G (7) 作用機序in vitro試験成績一覧(経口剤中の不純物および代謝物の薬理作用)
試験項目 試験法 試験系 試験結果 試験番号 経口剤(軟カプセル
剤)中の不純物およ
び代謝物のヒトオピ
オイド受容体作動性
フォルスコリン刺激
誘発 cAMP 産生に
対する抑制作用 使用濃度 nmol/L 10α-OH κ: 0.0003~100 μ, δ: 0.01~3000 de-CPM, NFA-G, de-CPM-G κ, μ, δ: 0.01~3000 TRK-820 κ: 0.00003~10 μ, δ: 0.01~3000 U-69593 κ: 0.001~300 DAMGO μ: 0.003~1000 DPDPE δ: 0.0003~100
ヒトオピオイド受
容体強制発現細胞 κ: CHO-K1 μ: CHO-dhfr(-) δ: CHO-dhfr(-)
被験物質 κ 受容体 μ 受容体 δ 受容体
EC50 (nmol/L)
Imax (%)
EC50 (nmol/L)
Imax (%)
EC50 (nmol/L)
Imax (%)
10α-OH (不純物)
0.0652 ± 0.0021
90.4 ± 0.7 N.C. (46.4
± 3.4)b N.C.a 16.6 ± 1.2
de-CPM (代謝物)
2.56 ± 0.14
90.6 ± 0.5 N.C. (70.4
± 1.5)b N.C. (41.6
± 2.4)b
NFA-G (代謝物)
43.2 ± 3.1
89.7 ± 0.6 N.C.a 14.1
± 3.6 N.C.a 6.6 ± 1.5
de-CPM-G (代謝物) N.C. (21.7
± 2.9)b N.C.a -5.5 ± 4.9 N.C.a 3.1
± 1.1
TRK-820 0.00940 ± 0.00138
90.8 ± 0.6
2.35 ± 0.31
50.3 ± 1.8
19.7 ± 1.3
78.5 ± 1.4
完全作動薬 c 0.525 ± 0.030
89.4 ± 0.7
6.49 ± 0.50
76.7 ± 1.1
0.119 ± 0.006
86.4 ± 0.3
数値は平均値 ± 標準誤差(n=4)を示す。 N.C.: 最高濃度において最大反応に達していなかったため、算出しなかった。 a: 最高濃度で最大反応に達していたが、作用が弱く、算出不能であった。 b: 最高濃度において最大反応に達していなかったため、参考値として最高濃
度での反応率を示した。 c: U-69593(オピオイド κ 受容体)、DAMGO(オピオイド μ 受容体)、
DPDPE(オピオイド δ 受容体) 結論: 経口剤中の不純物および代謝物のオピオイド κ、μ お
よび δ 受容体に対する作動性は TRK-820 と比較して
極めて弱いことが示唆された。最も活性が高かった
不純物 10α-OH においてもオピオイド κ 受容体に対
する作動性は TRK-820 の 1/7 であった。
- 110 -

ナルフラフィン塩酸塩 2.6.3 薬理試験概要表 Page 26
2.6.3.2 効力を裏付ける試験
被験物質: de-CPM、NFA-G、de-CPM-G (8) 作用機序in vitro試験成績一覧(代謝物の薬理作用)
試験項目 試験法 試験系 試験結果 試験番号 代謝物のオピオイド
受容体以外の受容
体、イオンチャネル
またはトランスポー
ターに対する作用
各受容体、イオ
ンチャネルおよ
びトランスポー
ターへの結合試
験 使用濃度 1000 nmol/L
各種組織ま
たは細胞
受容体 イオンチャネル
トランスポーター
阻害率 (%) de-CPM
1000 nmol/L NFA-G
1000 nmol/L de-CPM-G
1000 nmol/L Adenosine A1 2 -8 0 Adenosine A2A 7 0 3 Transporter, Adenosine -1 1 3 Adrenergic α1, Non-Selective 0 -4 2 Adrenergic α2, Non-Selective 9 14 12 Adrenergic β1 -2 1 4 Adrenergic β2 9 4 5 Transporter, Norepinephrine (NET) 6 6 3 Angiotensin AT2 -2 -2 -2 Atrial Natriuretic Factor (ANF) -2 2 -4 Bombesin, Non-Selective 8 17 8 Bradykinin B2 4 10 17 Calcitonin Gene-Related Peptide CGRP1
2 -2 4
Calcium Channel L-Type, Dihydropyridine 18 13 23
Calcium Channel N-Type 2 -5 2 Cannabinoid CB1 2 -14 2 Chemokine CCR1 2 -2 -1 Chemokine CCR2B -6 -4 -6 Cholecystokinin CCK1 (CCKA) 6 -5 11 Cholecystokinin CCK2 (CCKB) -5 -2 1 Transporter, Choline 17 9 1 Corticotropin Releasing Factor CRF1
-11 -5 1
Dopamine D1 -1 -9 -3 Dopamine D2L 3 -3 1 Dopamine D3 5 1 15 Transporter, Dopamine (DAT) -3 1 3
阻害率(%)は 1 回の実験(n=2)から算出した。
- 111 -
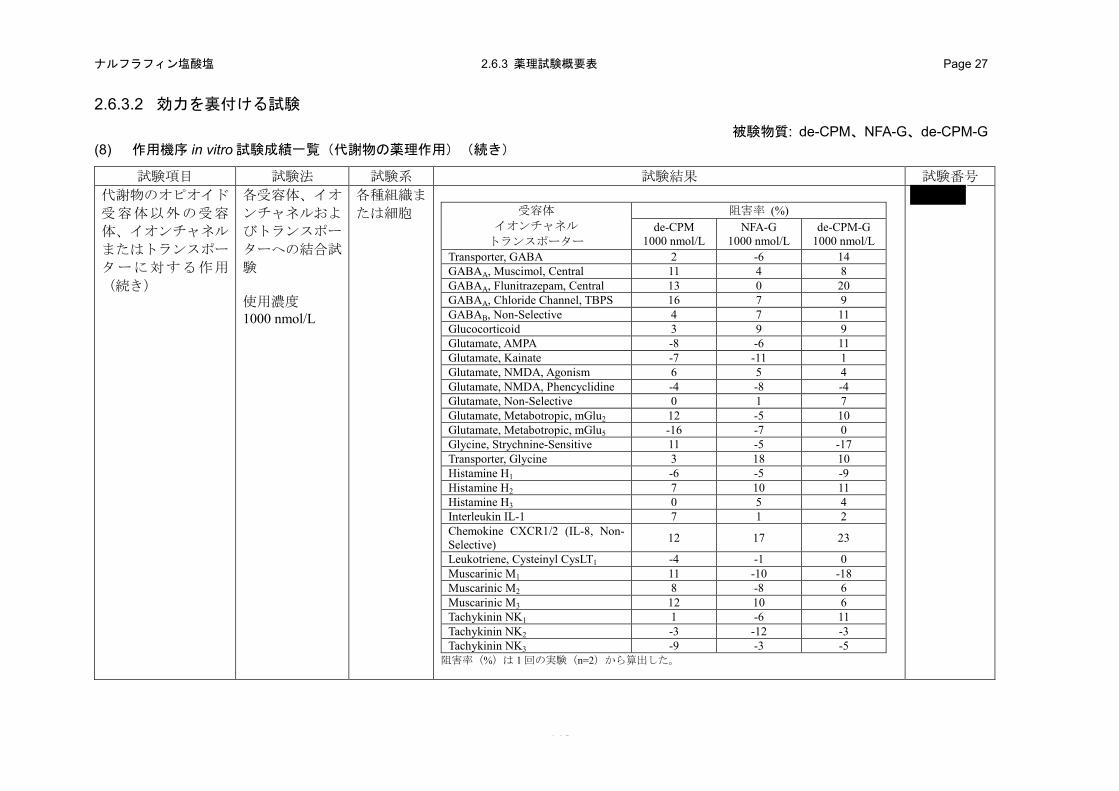
ナルフラフィン塩酸塩 2.6.3 薬理試験概要表 Page 27
2.6.3.2 効力を裏付ける試験
被験物質: de-CPM、NFA-G、de-CPM-G (8) 作用機序 in vitro 試験成績一覧(代謝物の薬理作用)(続き)
試験項目 試験法 試験系 試験結果 試験番号 代謝物のオピオイド
受容体以外の受容
体、イオンチャネル
またはトランスポー
ターに対する作用
(続き)
各受容体、イオ
ンチャネルおよ
びトランスポー
ターへの結合試
験 使用濃度 1000 nmol/L
各種組織ま
たは細胞
受容体 イオンチャネル
トランスポーター
阻害率 (%) de-CPM
1000 nmol/L NFA-G
1000 nmol/L de-CPM-G
1000 nmol/L Transporter, GABA 2 -6 14 GABAA, Muscimol, Central 11 4 8 GABAA, Flunitrazepam, Central 13 0 20 GABAA, Chloride Channel, TBPS 16 7 9 GABAB, Non-Selective 4 7 11 Glucocorticoid 3 9 9 Glutamate, AMPA -8 -6 11 Glutamate, Kainate -7 -11 1 Glutamate, NMDA, Agonism 6 5 4 Glutamate, NMDA, Phencyclidine -4 -8 -4 Glutamate, Non-Selective 0 1 7 Glutamate, Metabotropic, mGlu2 12 -5 10 Glutamate, Metabotropic, mGlu5 -16 -7 0 Glycine, Strychnine-Sensitive 11 -5 -17 Transporter, Glycine 3 18 10 Histamine H1 -6 -5 -9 Histamine H2 7 10 11 Histamine H3 0 5 4 Interleukin IL-1 7 1 2 Chemokine CXCR1/2 (IL-8, Non-Selective) 12 17 23
Leukotriene, Cysteinyl CysLT1 -4 -1 0 Muscarinic M1 11 -10 -18 Muscarinic M2 8 -8 6 Muscarinic M3 12 10 6 Tachykinin NK1 1 -6 11 Tachykinin NK2 -3 -12 -3 Tachykinin NK3 -9 -3 -5
阻害率(%)は 1 回の実験(n=2)から算出した。
- 112 -

ナルフラフィン塩酸塩 2.6.3 薬理試験概要表 Page 28
2.6.3.2 効力を裏付ける試験
被験物質: de-CPM、NFA-G、de-CPM-G (8) 作用機序 in vitro 試験成績一覧(代謝物の薬理作用)(続き)
試験項目 試験法 試験系 試験結果 試験番号 代謝物のオピオイド
受容体以外の受容
体、イオンチャネル
またはトランスポー
ターに対する作用
(続き)
各受容体、イオ
ンチャネルおよ
びトランスポー
ターへの結合試
験 使用濃度 1000 nmol/L
各種組織ま
たは細胞
受容体 イオンチャネル
トランスポーター
阻害率 (%) de-CPM
1000 nmol/L NFA-G
1000 nmol/L de-CPM-G
1000 nmol/L Neuropeptide Y Y2 -4 1 3 Nicotinic Acetylcholine 9 2 -5 Orphanin ORL1 3 -5 5 Orexin OX1 4 4 6 Platelet Activating Factor (PAF) -1 -7 -6 Potassium Channel [KA] 0 7 -1 Potassium Channel [KATP] 2 -3 3 Purinergic P2X 12 0 -4 Purinergic P2Y 18 22 31 Ryanodine RyR3 0 2 0 Serotonin 5-HT1, Non-Selective 6 -1 1 Serotonin 5-HT1A -3 -7 6 Serotonin 5-HT2, Non-Selective 0 1 13 Serotonin 5-HT2A 1 -2 -1 Serotonin 5-HT3 -5 0 4 Serotonin 5-HT4 23 -9 15 Transporter, Serotonin (SERT) 5 -9 -3 Sigma, Non-Selective -8 -5 -4 Somatostatin sst1 3 1 -4 Tumor Necrosis Factor (TNF), Non-Selective 5 9 9
Vasoactive Intestinal Peptide VIP1 -2 4 -3 Vasopressin V1A 4 5 -4 Vasopressin V2 3 -4 4
阻害率(%)は 1 回の実験(n=2)から算出した。 結論: TRK-820 の代謝物のオピオイド受容体以外の受容体、イオンチャネ
ルおよびトランスポーターに対する結合性は、TRK-820 のオピオイ
ド κ 受容体に対する結合性(2.6.3.2(1)、4.2.1.1-3: 試験)
と比較すると極めて低かった。
- 113 -

ナルフラフィン塩酸塩 2.6.3 薬理試験概要表 Page 29
2.6.3.2 効力を裏付ける試験
被験物質: TRK-820、de-CPM·T、NFA-G、de-CPM-G (9) 作用機序in vivo試験成績一覧(代謝物の薬理作用)
試験項目 試験目的 試験系 投与 経路 試験結果 試験番号
代謝物のサブスタンス P誘発引っ掻き行動に対
する作用
止痒作用発現に
おける代謝物の
関与を検討
ddY 系 雄性マウス
皮下 TRK-820(0.3、1、3 および 10 μg/kg)は用量依存的にサ
ブスタンス P(250 nmol/50 μL/site)皮内投与誘発引っ掻
き行動を抑制し、10 μg/kg で統計学的に有意な作用を示
した。ED50は 1.65 μg/kg(95%信頼区間: 0.880~3.08 μg/kg)であった。 de-CPM·T、NFA-G および de-CPM-G を TRK-820 が統計
学的に有意な抑制作用を示した 10 μg/kg の 100 倍量
(1、10、100 および 1000 μg/kg)まで投与しても統計学
的に有意な引っ掻き行動抑制作用は観察されなかった。 結論: 全身性に投与された TRK-820 の代謝物は止痒作用
を発現しない可能性が示唆されたことから、経口
投与による TRK-820 の止痒作用発現に代謝物は寄
与していないことが考えられた。
- 114 -

ナルフラフィン塩酸塩 2.6.3 薬理試験概要表 Page 30
2.6.3.3 副次的薬理試験
該当なし。
- 115 -

ナルフラフィン塩酸塩 2.6.3 薬理試験概要表 Page 31
2.6.3.4 安全性薬理試験
被験物質: TRK-820 (1) 安全性薬理コアバッテリー試験
評価対象となる組織 動物種/系統 投与 方法 投与量 a 性別および
動物数/群 特記すべき所見 GLP 適用 試験番号
1) 中枢神経系におよ
ぼす影響
a) ラットの一般症
状および行動に
およぼす影響 (Irwin 法)
ラット/Wistar 経口 100, 300, 1000, 3000 μg/kg
雄 n=6
100 および 300 μg/kg: 作用なし 1000 μg/kg: 身づくろいの軽度な低下、自発
運動の軽度な低下、体温の軽度な低下、軽
度な眼瞼下垂 3000 μg/kg: 警戒性の軽度な低下、身づくろ
いの軽度な低下、反応性の軽度な低下、自
発運動の軽度な低下、軽度なよろめき歩
行、軽度な眼瞼下垂、体温の軽度な低下な
いし低下、軽度な四肢伸長、軽度な流涙、
逃避反応の軽度な低下、痛覚反応の軽度な
低下、耳介反射の軽度な低下、軽度な腹這
い姿勢、軽度な異常歩行、軽度な縮瞳
否
a: 特に断りがない限り単回投与
- 116 -

ナルフラフィン塩酸塩 2.6.3 薬理試験概要表 Page 32
2.6.3.4 安全性薬理試験
被験物質: TRK-820 (1) 安全性薬理コアバッテリー試験(続き)
評価対象となる組織 動物種/系統 投与 方法 投与量 a 性別および
動物数/群 特記すべき所見 GLP 適用 試験番号
2) 心血管系におよぼ
す影響
a) hERG 電流に対
する作用(ホー
ルセルパッチク
ランプ法)
hERG 導入 HEK-293 細胞
in vitro 0, 3, 30, 300, 3000 nmol/L
n=5 0 nmol/L の抑制率: 4.9 ± 1.6%(平均値 ± 標準誤差)
3 nmol/L: 作用なし(4.9 ± 1.9%) 30 nmol/L: 抑制(12.1 ± 2.0%) 300 nmol/L: 抑制(27.2 ± 1.3%) 3000 nmol/L: 抑制(81.4 ± 1.2%)
IC50=840 nmol/L
適
b) 覚醒イヌを用い
た試験(無麻
酔・非拘束)
イヌ/ビーグル 経口 0, 3, 10, 100, 300 μg/kg
雄 n=4
3 μg/kg: 影響なし 10 μg/kg: 収縮期血圧、平均血圧の低下、心
拍数の増加 100 および 300 μg/kg: 収縮期血圧、平均血圧
の軽度な低下、心拍数の増加 心電図 (PR 間隔、QRS 時間、QT 間隔、
QTc): 影響なし
適
3) 呼吸系におよぼす
影響
a) 覚醒イヌを用い
た試験(無麻
酔・非拘束)
イヌ/ビーグル 経口 0, 3, 10, 100, 300 μg/kg
雄 n=4
呼吸数、pCO2 (mmHg)、pO2 (mmHg)、pH、
ヘモグロビン O2 saturation (%): 影響なし 適
a: 特に断りがない限り単回投与
- 117 -

ナルフラフィン塩酸塩 2.6.3 薬理試験概要表 Page 33
2.6.3.4 安全性薬理試験
被験物質: TRK-820 (2) 安全性薬理コアバッテリーに対するフォローアップ試験
評価対象となる組織 動物種/系統 投与 方法 投与量 a 性別および
動物数/群 特記すべき所見 GLP 適用 試験番号
1) 中枢神経系におよ
ぼす影響
a) サルの一般症状
および行動にお
よぼす影響
アカゲザル 静脈内 0.25, 0.5, 1, 2 μg/kg
雌雄 n=2
ただし、溶
媒投与はn=6
5%マンニトール(溶媒): 5 例で観察者への
攻撃行動の増加もしくは減弱、その内 1 例
で観察者への怯え表情の減弱 0.25 μg/kg: 作用なし 0.5 μg/kg: 2 例で運動低下、うずくまり姿
勢、観察者への攻撃行動の増強、1 例で観
察者への攻撃行動の減弱、腹臥位、動作緩
慢 1 μg/kg: 2 例で観察者への攻撃行動の減弱、
1 例で運動低下、うずくまり姿勢、腹臥
位、観察者への怯え表情の減弱、動作緩
慢、閉眼 2 μg/kg: 2 例で流涎、運動低下、うずくまり
姿勢、閉眼、動作緩慢、口の半開状態、 1 例で観察者への攻撃行動の減弱、腹臥
位、運動失調
適
a: 特に断りがない限り単回投与
- 118 -

ナルフラフィン塩酸塩 2.6.3 薬理試験概要表 Page 34
2.6.3.4 安全性薬理試験
被験物質: TRK-820 (2) 安全性薬理コアバッテリーに対するフォローアップ試験(続き)
評価対象となる組織 動物種/系統 投与 方法 投与量 a 性別および
動物数/群 特記すべき所見 GLP 適用 試験番号
1) 中枢神経系におよ
ぼす影響
b) 自発運動に対す
る作用
移所運動 マウス/ICR 経口 50, 100, 200, 400 μg/kg
雄 n=7~8
400 μg/kg: 有意な抑制作用あり ED50: 344.8 μg/kg
(95%信頼区間: 258.2~627.4 μg/kg)
否
ラット/Wistar 経口 100, 300, 1000, 3000 μg/kg
雄 n=6
1000 μg/kg 以上: 有意な抑制作用あり 否
a: 特に断りがない限り単回投与
- 119 -

ナルフラフィン塩酸塩 2.6.3 薬理試験概要表 Page 35
2.6.3.4 安全性薬理試験
被験物質: TRK-820 (2) 安全性薬理コアバッテリーに対するフォローアップ試験(続き)
評価対象となる組織 動物種/系統 投与 方法 投与量 a 性別および
動物数/群 特記すべき所見 GLP 適用 試験番号
1) 中枢神経系におよ
ぼす影響
b) 自発運動に対す
る作用
回転かご マウス/ICR 経口 10, 30, 100, 300 μg/kg
雄 n=6~7
30 および 60 分間測定 100 μg/kg 以上: 有意な抑制作用あり 30 分間測定
ED50: 103 μg/kg (95%信頼区間: 64.9~163 μg/kg)
60 分間測定
ED50: 90.6 μg/kg (95%信頼区間: 54.5~151 μg/kg)
否
マウス/ddY 皮下 1, 3, 10, 30 μg/kg
雄 n=6~8
60 分間測定 10 μg/kg 以上: 有意な抑制作用あり ED50: 7.79 μg/kg
(95%信頼区間: 3.82~15.9 μg/kg)
否
a: 特に断りがない限り単回投与
- 120 -

ナルフラフィン塩酸塩 2.6.3 薬理試験概要表 Page 36
2.6.3.4 安全性薬理試験
被験物質: TRK-820 (2) 安全性薬理コアバッテリーに対するフォローアップ試験(続き)
評価対象となる組織 動物種/系統 投与 方法 投与量 a 性別および
動物数/群 特記すべき所見 GLP 適用 試験番号
1) 中枢神経系におよ
ぼす影響
c) 協調運動に対す
る作用(ロタロ
ッド)
マウス/ddY 経口 10, 30, 100, 300 μg/kg
雄 n=10
投与 30 分後 300 μg/kg: 有意な影響あり ED50: 115 μg/kg
(95%信頼区間: 68.6~194 μg/kg) 投与 60 分後
100 μg/kg 以上: 有意な影響あり ED50: 72.3 μg/kg
(95%信頼区間: 44.0~119 μg/kg)
否
d) 麻酔作用(ペン
トバルビタール
誘発睡眠)
マウス/ICR 経口 30, 100, 300, 1000, 3000 μg/kg
雄 n=8
1000 μg/kg 以上: 睡眠時間の有意な延長作用
あり 適
e) 脳波に対する作
用(無麻酔·無拘
束下·自発脳波)
ラット/Wistar 皮下 3, 10, 30 μg/kg
雄 n=6
脳波の波形に対する影響なし 新皮質前頭葉周波数解析
3 μg/kg: アルファ帯域の減少 10 μg/kg 以上: アルファ、ベータ-1 帯域の
減少 海馬周波数解析
10 μg/kg: アルファ帯域の減少、ベータ-2 帯
域の増加 30 μg/kg: アルファ帯域の減少
適
a: 特に断りがない限り単回投与
- 121 -
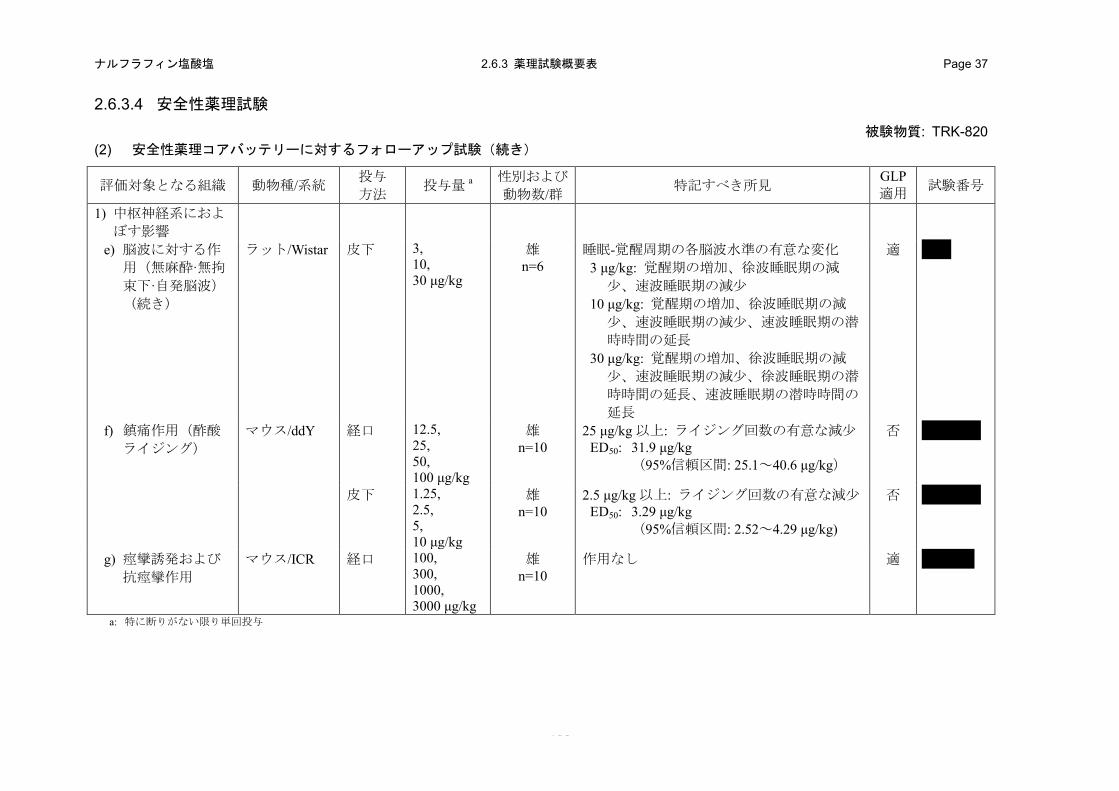
ナルフラフィン塩酸塩 2.6.3 薬理試験概要表 Page 37
2.6.3.4 安全性薬理試験
被験物質: TRK-820 (2) 安全性薬理コアバッテリーに対するフォローアップ試験(続き)
評価対象となる組織 動物種/系統 投与 方法 投与量 a 性別および
動物数/群 特記すべき所見 GLP 適用 試験番号
1) 中枢神経系におよ
ぼす影響
e) 脳波に対する作
用(無麻酔·無拘
束下·自発脳波)
(続き)
ラット/Wistar 皮下 3, 10, 30 μg/kg
雄 n=6
睡眠-覚醒周期の各脳波水準の有意な変化 3 μg/kg: 覚醒期の増加、徐波睡眠期の減
少、速波睡眠期の減少 10 μg/kg: 覚醒期の増加、徐波睡眠期の減
少、速波睡眠期の減少、速波睡眠期の潜
時時間の延長 30 μg/kg: 覚醒期の増加、徐波睡眠期の減
少、速波睡眠期の減少、徐波睡眠期の潜
時時間の延長、速波睡眠期の潜時時間の
延長
適
f) 鎮痛作用(酢酸
ライジング) マウス/ddY 経口 12.5,
25, 50, 100 μg/kg
雄 n=10
25 μg/kg 以上: ライジング回数の有意な減少 ED50: 31.9 μg/kg
(95%信頼区間: 25.1~40.6 μg/kg)
否
皮下 1.25, 2.5, 5, 10 μg/kg
雄 n=10
2.5 μg/kg 以上: ライジング回数の有意な減少 ED50: 3.29 μg/kg
(95%信頼区間: 2.52~4.29 μg/kg)
否
g) 痙攣誘発および
抗痙攣作用 マウス/ICR 経口 100,
300, 1000, 3000 μg/kg
雄 n=10
作用なし 適
a: 特に断りがない限り単回投与
- 122 -

ナルフラフィン塩酸塩 2.6.3 薬理試験概要表 Page 38
2.6.3.4 安全性薬理試験
被験物質: TRK-820 (2) 安全性薬理コアバッテリーに対するフォローアップ試験(続き)
評価対象となる組織 動物種/系統 投与 方法 投与量 a 性別および
動物数/群 特記すべき所見 GLP 適用 試験番号
1) 中枢神経系におよ
ぼす影響
h) 正常体温に対す
る作用(直腸
温)
ラット/Wistar 経口 100, 300, 1000, 3000 μg/kg
雄 n=6
1000 μg/kg: 体温の軽度な低下(溶媒対照群
の体温の-0.3~-0.5°C) 3000 μg/kg: 体温の低下(溶媒対照群の体温
の-1.2~-1.5°C)
否
2) 心血管系におよぼ
す影響
a) 摘出乳頭筋の活
動電位に対する
作用
モルモット/ Hartley
in vitro 0, 30, 300, 3000 nmol/L
雄 n=6
0 nmol/L: APD50: 100.9 ± 1.2%、APD90: 99.9 ± 1.0%(平均値 ± 標準偏差)
30 nmol/L: 作用なし(APD50: 99.0 ± 2.4%、APD90: 98.7 ± 1.9%)
300 nmol/L: 作用なし(APD50: 102.9 ± 0.9%、APD90: 103.5 ± 0.9%)
3000 nmol/L: 延長(APD50: 114.1 ± 6.1%、APD90: 116.4 ± 6.1%)
静止膜電位、活動電位振幅、APD20、最大立
ち上がり速度には作用なし
適
a: 特に断りがない限り単回投与
- 123 -
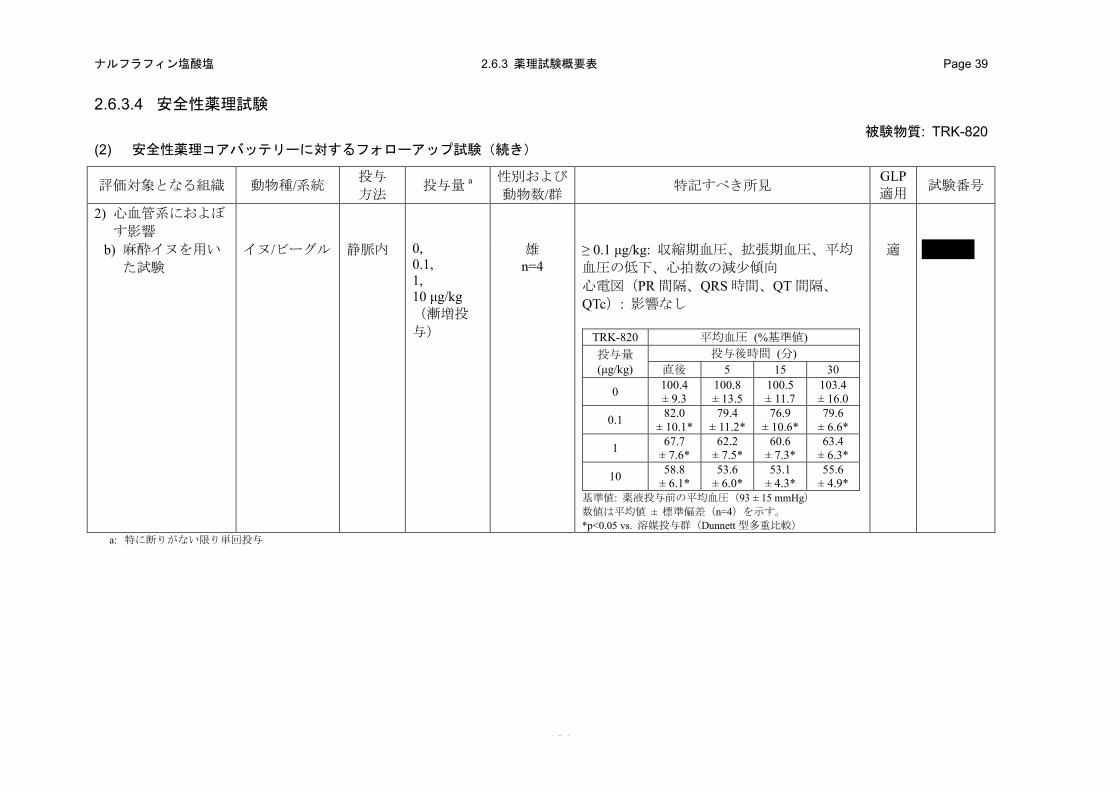
ナルフラフィン塩酸塩 2.6.3 薬理試験概要表 Page 39
2.6.3.4 安全性薬理試験
被験物質: TRK-820 (2) 安全性薬理コアバッテリーに対するフォローアップ試験(続き)
評価対象となる組織 動物種/系統 投与 方法 投与量 a 性別および
動物数/群 特記すべき所見 GLP 適用 試験番号
2) 心血管系におよぼ
す影響
b) 麻酔イヌを用い
た試験 イヌ/ビーグル 静脈内 0,
0.1, 1, 10 μg/kg (漸増投
与)
雄 n=4
≥ 0.1 μg/kg: 収縮期血圧、拡張期血圧、平均
血圧の低下、心拍数の減少傾向 心電図(PR 間隔、QRS 時間、QT 間隔、
QTc): 影響なし
TRK-820 平均血圧 (%基準値) 投与量(μg/kg)
投与後時間 (分) 直後 5 15 30
0 100.4 ± 9.3
100.8 ± 13.5
100.5 ± 11.7
103.4 ± 16.0
0.1 82.0 ± 10.1*
79.4 ± 11.2*
76.9 ± 10.6*
79.6 ± 6.6*
1 67.7 ± 7.6*
62.2 ± 7.5*
60.6 ± 7.3*
63.4 ± 6.3*
10 58.8 ± 6.1*
53.6 ± 6.0*
53.1 ± 4.3*
55.6 ± 4.9*
基準値: 薬液投与前の平均血圧(93 ± 15 mmHg) 数値は平均値 ± 標準偏差(n=4)を示す。 *p<0.05 vs. 溶媒投与群(Dunnett 型多重比較)
適
a: 特に断りがない限り単回投与
- 124 -

ナルフラフィン塩酸塩 2.6.3 薬理試験概要表 Page 40
2.6.3.4 安全性薬理試験
被験物質: TRK-820 (3) 補足的安全性薬理試験
評価対象となる組織 動物種/系統 投与 方法 投与量 a 性別および
動物数/群 特記すべき所見 GLP 適用 試験番号
1) 腎/泌尿器系にお
よぼす影響 ラット/SD 経口 3,
10, 30, 100, 300, 1000 μg/kg
雄 n=6
10 μg/kg: 尿中 Na+排泄量の有意な減少 (29%減少)
100 μg/kg: 尿中 Na+排泄量の有意な減少 (34%減少)
300 μg/kg: 尿量の有意な増加(292%増加)、
尿中 K+排泄量の有意な増加(52%増加) 1000 μg/kg: 尿量の有意な増加(408%増加)、
尿中 Na+排泄量の有意な減少(45%減少)、
尿中 K+排泄量の有意な増加(54%増加)、
尿中 Cl–排泄量の有意な減少(38%減少)
適
2) 自律神経系にお
よぼす影響(摘
出回腸)
モルモット/ Hartley
in vitro 100, 300, 1000 nmol/L
雄 n=4
作用なし 適
3) 胃腸管系におよ
ぼす影響(腸管
輸送能)
マウス/ICR 経口 100, 300, 1000, 3000 μg/kg
雄 n=8
1000 μg/kg: 有意に抑制 (抑制率 31.5 ± 7.59%)
3000 μg/kg: 有意に抑制 (抑制率 52.9 ± 4.63%)
ED50: 3470 μg/kg (95%信頼区間: 1620~19400 μg/kg)
抑制率は平均値 ± 標準誤差を示す
否
a: 特に断りがない限り単回投与
- 125 -

ナルフラフィン塩酸塩 2.6.3 薬理試験概要表 Page 41
2.6.3.5 薬力学的薬物相互作用試験
被験物質: TRK-820 (1) in vivo試験成績一覧
試験項目 試験目的 試験系 投与 経路 試験結果 試験番号
1) 抗ヒスタミン薬
ケトチフェンと
の相互作用
ペントバルビタール
誘発睡眠時間の延長
作用を指標に中枢抑
制作用における相互
作用を検討
ICR 系 雄性マウス
皮下
TRK-820 (μg/kg, 皮下)
ケトチフェン
(mg/kg, 経口)
ペントバルビタール (50 mg/kg, 腹腔内) 誘発睡眠時間(分)
0 0 32.1 ± 2.51 10 0 28.2 ± 3.24 100 0 54.6 ± 4.40** 0 20 61.2 ± 8.61#
10 20 52.7 ± 4.16 100 20 80.7 ± 6.54
数値は平均値 ± 標準誤差(n=8)を示す。 **p<0.01 vs. TRK-820 の溶媒/ケトチフェンの溶媒投与群(Dunnett 型多重比較) #p<0.05 vs. TRK-820 の溶媒/ケトチフェンの溶媒投与群(Welch 検定) ケトチフェン単独投与群と TRK-820/ケトチフェン併用投与群との間で Dunnett 型多重比較を実施したが有意な差は検出できなかった。 結論: 単独ではペントバルビタール誘発睡眠時間の延長作用
を示さない TRK-820 の 10 μg/kg をケトチフェンと併用
投与しても睡眠時間はケトチフェン単独投与群と同程
度であった。一方、単独で睡眠時間の延長作用を示す
TRK-820 の 100 μg/kg をケトチフェンと併用投与する
と、睡眠時間はケトチフェン単独投与群よりも 20 分延
長したことから、中枢抑制作用において TRK-820 とケ
トチフェンが相加的に作用する可能性が推察された。
- 126 -

ナルフラフィン塩酸塩 2.6.3 薬理試験概要表 Page 42
2.6.3.5 薬力学的薬物相互作用試験
被験物質: TRK-820 (1) in vivo 試験成績一覧(続き)
試験項目 試験目的 試験系 投与 経路 試験結果 試験番号
2) 睡眠導入薬ニト
ラゼパムとの相
互作用
ペントバルビタール
誘発睡眠時間の延長
作用を指標に中枢抑
制作用における相互
作用を検討
ICR 系 雄性マウス
皮下
TRK-820 (μg/kg, 皮下)
ニトラゼパム (mg/kg, 腹腔内)
ペントバルビタール (50 mg/kg, 腹腔内) 誘発睡眠時間(分)
0 0 31.4 ± 2.95 0 3 68.5 ± 4.35** 1 3 66.1 ± 6.21
10 3 91.1 ± 2.83## 100 3 96.8 ± 4.39##
数値は平均値 ± 標準誤差(n=10)を示す。 **p<0.01 vs. TRK-820 の溶媒/ニトラゼパムの溶媒投与群(t 検定) ##p<0.01 vs. TRK-820 の溶媒/ニトラゼパム投与群(Dunnett 型多重比較) 結論: TRK-820 の 1 μg/kg はニトラゼパムによるペントバルビ
タール誘発睡眠時間延長作用に影響しなかったが、
TRK-820 の 10 および 100 μg/kg をニトラゼパムと併用
投与すると、睡眠時間はニトラゼパム単独投与群より
も統計学的に有意に延長したことから、TRK-820 はニ
トラゼパムの睡眠作用を用量依存的に増強する可能性
が示唆された。
- 127 -


















