
2015-01-20 Modeling for Systems Pharmacologynkhuri/files/systems-pharmacology-lecture3... ·...
65
Molecular Modeling for Systems Pharmacology: Integrative modeling of biomolecular assemblies and networks 1/20/15 Andrej Sali ([email protected]) While it may be hard to live with generalization, it is inconceivable to live without it. Peter Gay, Schnitzler’s Century (2002).
Transcript of 2015-01-20 Modeling for Systems Pharmacologynkhuri/files/systems-pharmacology-lecture3... ·...

Molecular Modeling for Systems Pharmacology:
Integrative modeling of biomolecular assemblies and networks
1/20/15
Andrej Sali ([email protected])
While it may be hard to live with generalization, it is inconceivable to live without it. Peter Gay, Schnitzler’s Century (2002).

Contents
1. Hierarchy of biological organization and systems pharmacology
2. Modeling
3. Integrative modeling of assembly structures
4. Integrative modeling of molecular networks

Contents
1. Hierarchy of biological organization and systems pharmacology
2. Modeling
3. Integrative modeling of assembly structures
4. Integrative modeling of molecular networks

Hierarchy of biological organization
ORGANISM POPULATION
Representation: molecular structure (particles in space), localization (contents of volume elements), networks of components, inter-compartmental flows, …

Drug Discovery / Pharmacology
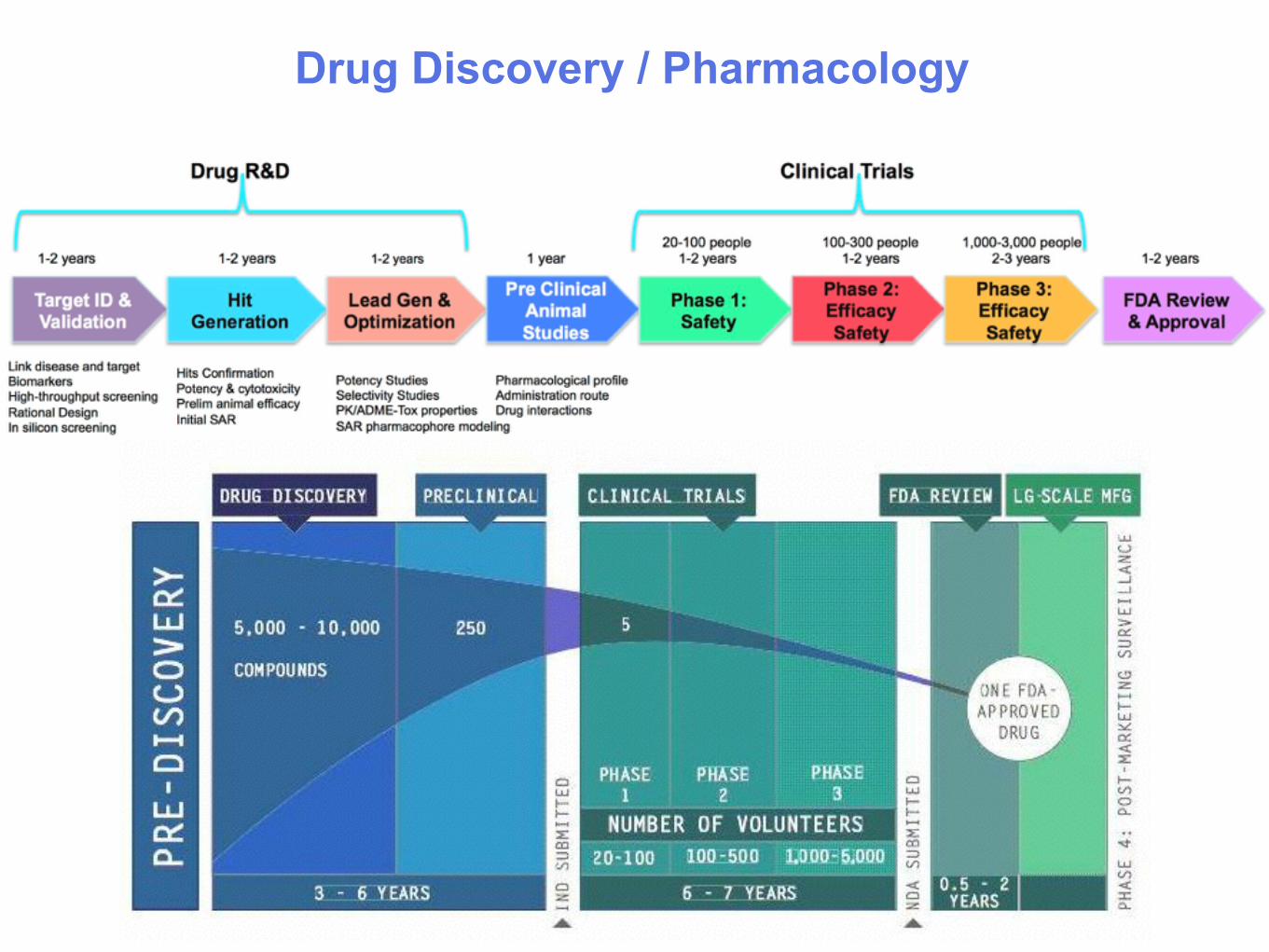
Drug Discovery / Pharmacology
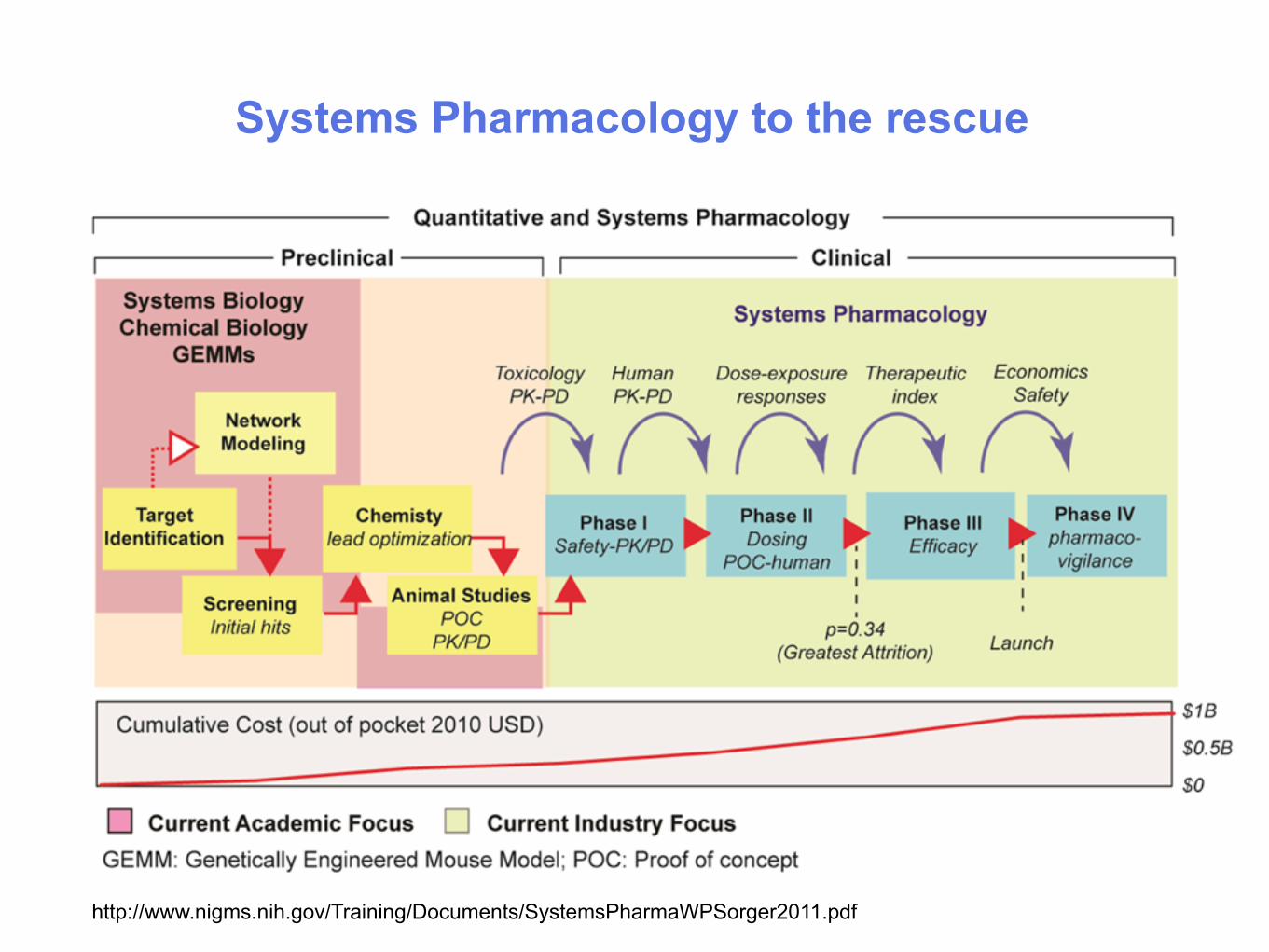
Systems Pharmacology to the rescue
http://www.nigms.nih.gov/Training/Documents/SystemsPharmaWPSorger2011.pdf

What is Systems Pharmacology?
1. Systems pharmacology is the application of systems biology principles to the field of pharmacology. It seeks to understand how medicines work on various systems of the body. Instead of considering the effect of a drug to be the result of one specific drug-protein interaction, systems pharmacology considers the effect of a drug to be the outcome of the network of interactions a drug may have. (Wikipedia)
2. Systems Pharmacology brings a new combination of mathematical and experimental tools to bear on the discovery and analysis of therapeutic drugs. System-level understanding of pharmacological effects will help to identify new uses for existing drugs, identify those patients most likely to benefit from mono and combination therapies, and make drug discovery and development faster, cheaper, and more predictable. (http://isp.hms.harvard.edu)

Systems Pharmacology Bibliography
1. Sorger, P. (2011). Quantitative and Systems Pharmacology in the Post-genomic Era: New Approaches to Discovering Drugs and Understanding Therapeutic Mechanisms (http://www.nigms.nih.gov/Training/Documents/SystemsPharmaWPSorger2011.pdf)
2. Dar, A. C., Das, T. K., Shokat, K. M., & Cagan, R. L. (2013). Chemical genetic discovery of targets and anti-targets for cancer polypharmacology. Nature, 486(7401), 80–84.
3. Csermely, P., Korcsmáros, T., Kiss, H. J. M., London, G., & Nussinov, R. (2013). Pharmacology & Therapeutics. Pharmacology and Therapeutics, 138(3), 333–408.
4. Zhao, S., & Iyengar, R. (2012). Systems Pharmacology: Network Analysis to Identify Multiscale Mechanisms of Drug Action. Annual Review of Pharmacology and Toxicology, 52(1), 505–521.
5. L Kapitzky, P Beltrao, TJ Berens, N Gassner, Ci Zhou, A Wuster, J Wu, MM Babu, SJ Elledge, D Toczyski, RS Lokey and NJ Krogan. Cross-species chemogenomic profiling reveals evolutionarily conserved drug mode of action. Molecular Systems Biology 6:451, 2010.
6. http://isp.hms.harvard.edu
7. http://www.nature.com/psp

Contents
1. Hierarchy of biological organization and systems pharmacology
2. Modeling
3. Integrative modeling of assembly structures
4. Integrative modeling of molecular networks

Scientific progress
1. Iteration of Hypothesis and Experiment
HypothesisModel
Experiment

Scientific progress
1. Iteration of Hypothesis and Experiment
HypothesisModel
Experiment
Hypothesis vs Theory

Scientific progress
1. Iteration of Hypothesis and Experiment
HypothesisModel
Experiment
Thomas Kuhn, 1962. The Structure of Scientific Revolutions
Hypothesis vs Theory

Scientific progress
1. Iteration of Hypothesis and Experiment
HypothesisModel
Experiment
Thomas Kuhn, 1962. The Structure of Scientific Revolutions
2. Exploratory research (eg, genome sequencing).
Hypothesis vs Theory

Modeling
Scientific modelling is a scientific activity the aim of which is to make a particular part or feature of the world easier to understand, define, quantify, visualize, or simulate. It requires selecting and identifying relevant aspects of a situation in the real world and then using different types of models for different aims, such as conceptual models to better understand, operational models to operationalize, mathematical models to quantify, and graphical models to visualize the subject. Modelling is an essential and inseparable part of scientific activity, and many scientific disciplines have their own ideas about specific types of modeling. (Wikipedia)


Composition Stoichiometry Chemical complementarity
X-ray diffraction

Composition Stoichiometry Chemical complementarity
X-ray diffraction
To understand and modulate cellular processes, we need their models.
These models are best generated by considering all available information.

1. Hierarchy of biological organization and systems pharmacology
2. Modeling
3. Integrative modeling of assembly structures
4. Integrative modeling of molecular networks
Contents

Structural biology: Maximize accuracy, resolution, completeness, and efficiency of the
structural coverage of macromolecular assemblies
Motivation: Models will allow us to understand how machines work, how they evolved, how they can be controlled, modified, and perhaps even designed.
There may be thousands of biologically relevant macromolecular complexes whose structures are yet to be characterized, involved in a few hundred core biological processes.
GroEL chaperonin
flagellar motorHIV virus
nuclear pore complexATP synthase ribosome
tRNA synthetaseRNA polymerase II

02/15/2007
Sali A, Earnest T, Glaeser R, Baumeister W. From words to literature in structural proteomics. Nature 422, 216-225, 2003. Ward A, Sali A, Wilson I. Integrative structural biology. Science 339, 913-915, 2013.
PHYSICS
STATISTICSEXPERIMENT
∫
Integrative Structural Biology for maximizing accuracy, resolution, completeness, and efficiency of structure determination
Use structural information from any source: measurement, first principles, rules; resolution: low or high resolution
to obtain the set of all models that are consistent with it.
INTUITION
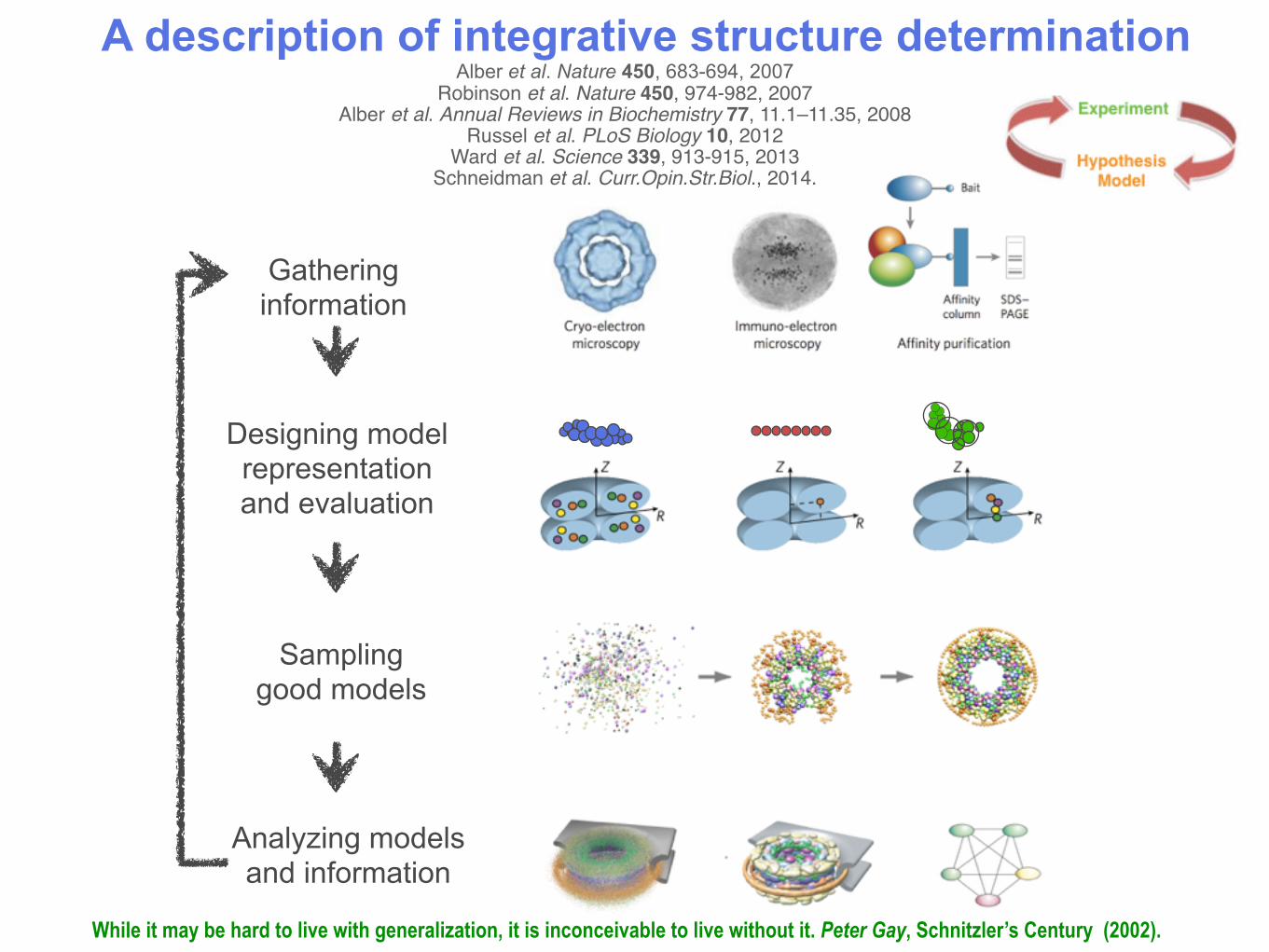
Gatheringinformation
Analyzing modelsand information
Samplinggood models
Designing modelrepresentationand evaluation
A description of integrative structure determinationAlber et al. Nature 450, 683-694, 2007
Robinson et al. Nature 450, 974-982, 2007Alber et al. Annual Reviews in Biochemistry 77, 11.1–11.35, 2008
Russel et al. PLoS Biology 10, 2012Ward et al. Science 339, 913-915, 2013
Schneidman et al. Curr.Opin.Str.Biol., 2014.
While it may be hard to live with generalization, it is inconceivable to live without it. Peter Gay, Schnitzler’s Century (2002).

Challenges in interpreting the data in terms of a structural model
• Sparseness due to incompleteness of measurements
• Error due to measurement and other imperfections
• Ambiguity eg, due to multiple copies of a protein in a system
• Incoherence (mixture) due to multiple states of a system in a heterogenous sample

Pushing the envelope of structural biology by integration of all available information
• Size
• Static systems in single and multiple states
• Dynamic systems
• Bulk and single molecule views
• Impure samples
• Overlapping with other domains such as systems biology
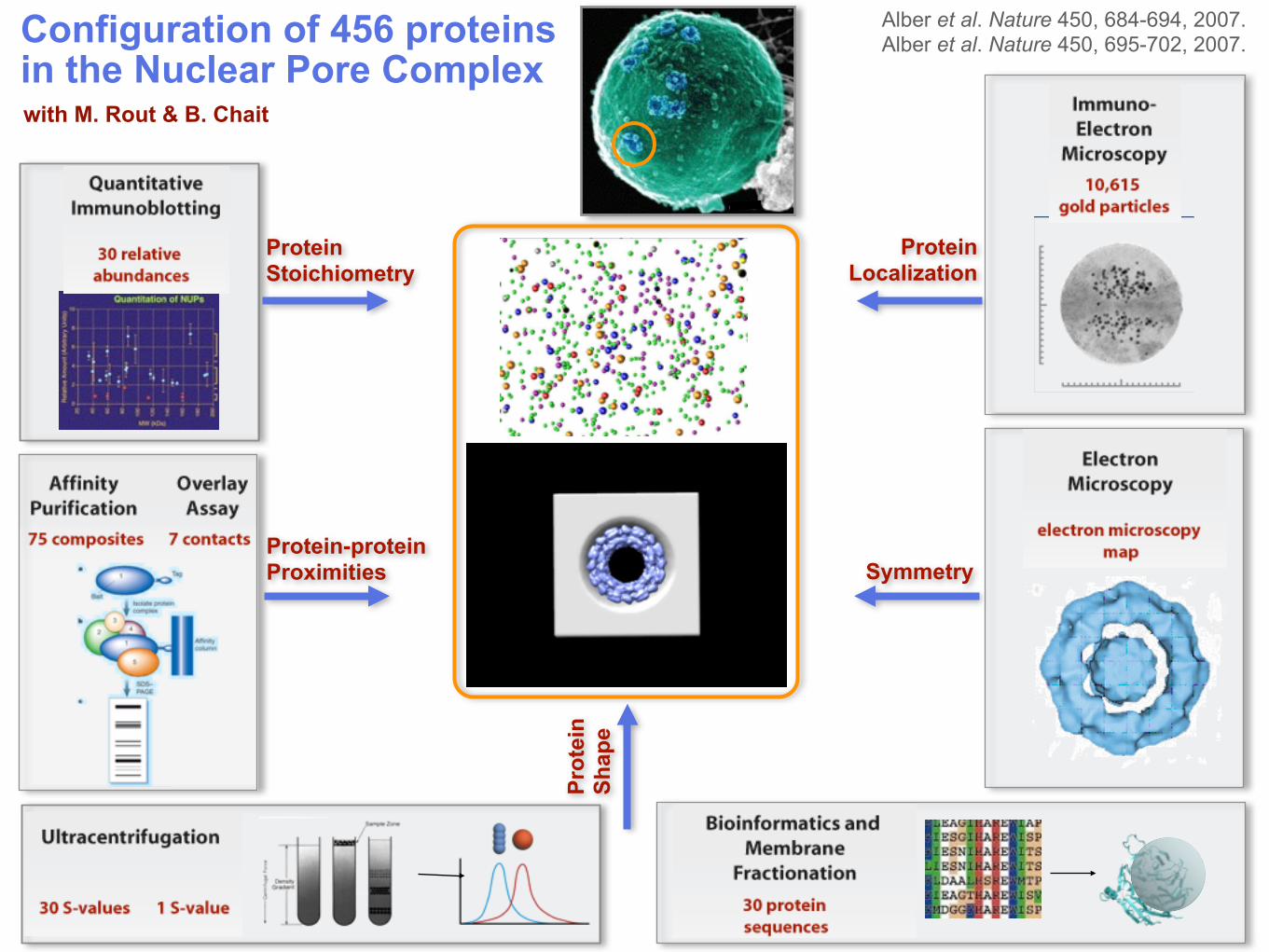
Prot
ein
Sh
ape
Protein Stoichiometry
Protein-protein Proximities Symmetry
Protein Localization
Configuration of 456 proteins in the Nuclear Pore Complex
Alber et al. Nature 450, 684-694, 2007. Alber et al. Nature 450, 695-702, 2007.
with M. Rout & B. Chait

Prot
ein
Sh
ape
Protein Stoichiometry
Protein-protein Proximities Symmetry
Protein Localization
Configuration of 456 proteins in the Nuclear Pore Complex
Alber et al. Nature 450, 684-694, 2007. Alber et al. Nature 450, 695-702, 2007.
with M. Rout & B. Chait

Open source, versions, documentation, wiki, examples, mailing lists, unit testing, bug tracking, ...
Integrative Modeling Platform (IMP) http://integrativemodeling.org
D. Russel, K. Lasker, B. Webb, J. Velazquez-Muriel, E. Tjioe, D. Schneidman, F. Alber, B. Peterson, A. Sali, PLoS Biol, 2012. R. Pellarin, M. Bonomi, B. Raveh, S. Calhoun, C. Greenberg, G.Dong.
IMP C++/Python library
restrainer
Simplicity
Flex
ibili
ty
Domain-specific
Chimera
Model
angle restraint
volume restraint
conjugate gradients
Monte Carlo
harmonic
nonbonded list
particledistance score
IO
connectivity restraint
cross correlation
Domino
rigid bodySAXS
docking
molecule
Representation: Atomic Rigid bodies Coarse-grained Multi-scale Symmetry / periodicity Multi-state systems
Scoring: Density maps EM images Proteomics FRET Chemical and Cys cross-linking Homology-derived restraints SAXS Native mass spectrometry Statistical potentials Molecular mechanics forcefields Bayesian scoring Library of functional forms (ambiguity, ...)
Analysis: Clustering Chimera Pymol PDB files Density maps
Sampling: Simplex Conjugate Gradients Monte Carlo Brownian Dynamics Molecular Dynamics Replica Exchange Divide-and-conquer enumeration

Modeling aspects
Sampling and scoring
1.Representation
2.Scoring function
3.Sampling algorithm
4.Assessment
Find simplest possible models that reproduce the measured data within their uncertainty.

Representation
1 5 10 20 40 60 100
Ignorance (residues per bead)
Hierarchical model representation facilitates using imprecise information

X structure d measured data point i f computed data point i (forward model) w weight of data point i
Scoring function: Least-squares modeling of a structure
S(X) = wii∑ [di − fi (X)]
2i
i
i
When , we get a “maximum likelihood” approach,
for which .
wi = 1/σ i2
S(X) = χ 2
S
ΔD

Scoring function: Bayesian modeling of structure(s)
Model M can include coordinates of one or more structures X as well as additional parameters (noise levels, weights, calibration parameters, ...).
likelihoodposterior prior
D � f(M)
Likelihood is the probability density of observing data D, given model M and prior information I (by relying on a model of noise and a forward model, which computes data D given model M).
Prior is the probability density of model M, given prior information I.
Posterior is the probability density of model M, given data D and information I.
Rieping, Habeck, Nilges. Science, 2005
M model D data I prior information f(M) forward model
p(AB) = p(BA) = p(A) ⋅ p(B/A) = p(B) ⋅ p(A/B)

Sampling, optimization, enumeration

Sampling, optimization, enumeration

Assessment(in the absence of Bayesian inference)

1.Existence of a good-scoring model.
Assessment(in the absence of Bayesian inference)

1.Existence of a good-scoring model.
2.Precision of the ensemble of good-scoring models.
Assessment(in the absence of Bayesian inference)

1.Existence of a good-scoring model.
2.Precision of the ensemble of good-scoring models.
3.Check model against unused data (cross-validation).
Assessment(in the absence of Bayesian inference)

1.Existence of a good-scoring model.
2.Precision of the ensemble of good-scoring models.
3.Check model against unused data (cross-validation).
4.Known precision / accuracy for “similar” cases.
Assessment(in the absence of Bayesian inference)

1.Existence of a good-scoring model.
2.Precision of the ensemble of good-scoring models.
3.Check model against unused data (cross-validation).
4.Known precision / accuracy for “similar” cases.
5.Non-random patterns in the model.
Assessment(in the absence of Bayesian inference)

1.Existence of a good-scoring model.
2.Precision of the ensemble of good-scoring models.
3.Check model against unused data (cross-validation).
4.Known precision / accuracy for “similar” cases.
5.Non-random patterns in the model.
Assessment
Modeling facilitates assessing the data as well as models in terms of precision and accuracy.
(in the absence of Bayesian inference)
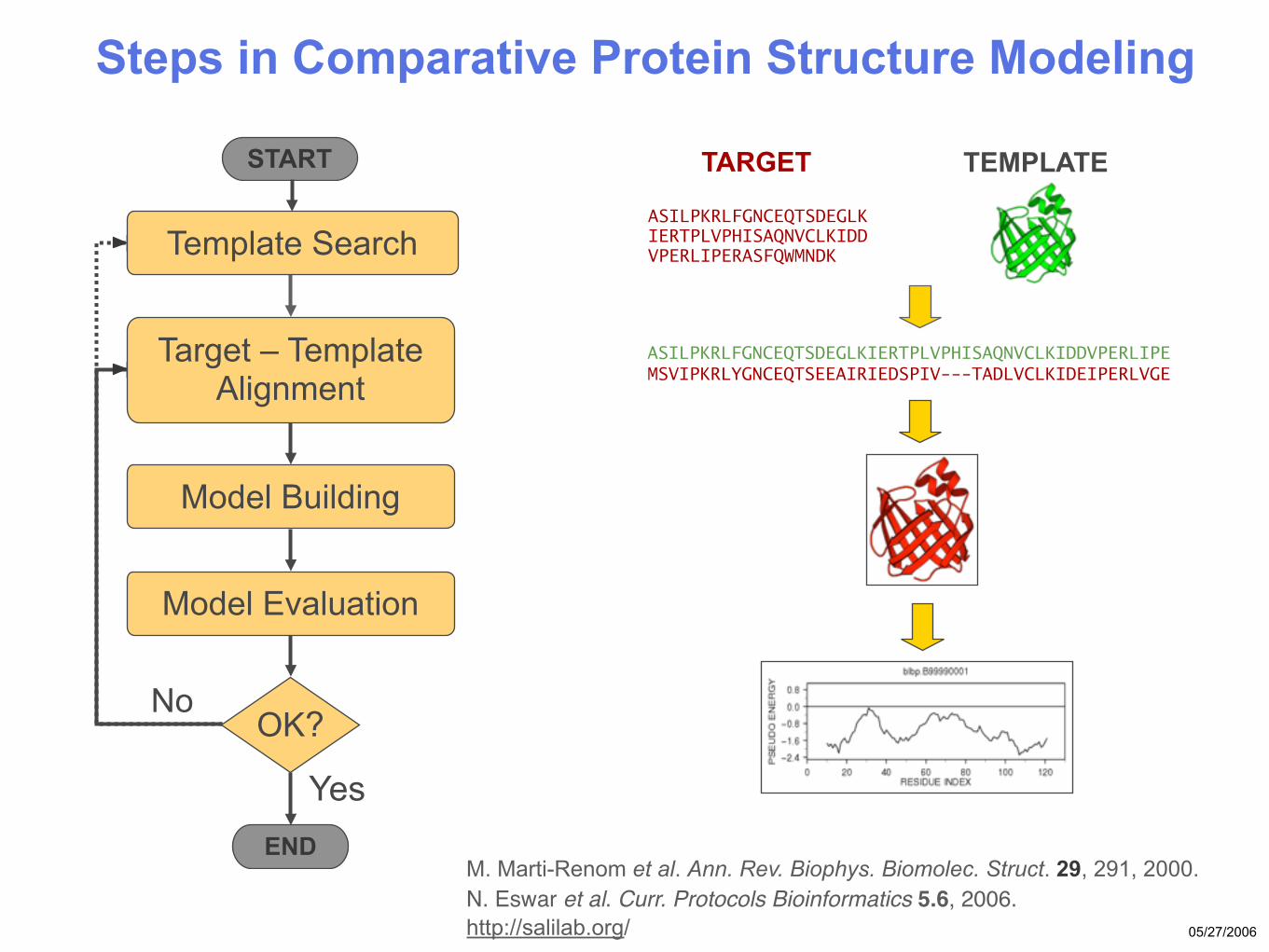
05/27/2006
Steps in Comparative Protein Structure Modeling
No
Target – Template Alignment MSVIPKRLYGNCEQTSEEAIRIEDSPIV---TADLVCLKIDEIPERLVGE
ASILPKRLFGNCEQTSDEGLKIERTPLVPHISAQNVCLKIDDVPERLIPE
Model Building
START
ASILPKRLFGNCEQTSDEGLKIERTPLVPHISAQNVCLKIDDVPERLIPERASFQWMNDK
TARGET
Template Search
TEMPLATE
OK?
Model Evaluation
END
Yes
M. Marti-Renom et al. Ann. Rev. Biophys. Biomolec. Struct. 29, 291, 2000. N. Eswar et al. Curr. Protocols Bioinformatics 5.6, 2006. http://salilab.org/

1. Hierarchy of biological organization and systems pharmacology
2. Modeling
3. Integrative modeling of assembly structures
4. Integrative modeling of molecular networks
Contents

Integrating structural biology and systems biology
∫CJ Ryan, P Cimermancic, ZA Szpiech, A Sali, RD Hernandez, NJ Krogan Nat Gen 14, 2013.

Annotation of enzyme function
1. Prediction of the function in the context of the pathway
2. Prediction of the pathway based on all available information
3. Automated (ie, thorough, objective, …) method

Computational Geometry Lab Tel-Aviv University, Israel
Mapping the Phase Space of Models for Transport through the NPC
What is the pathway, given a set of potential enzymes and metabolites as well as at least one enzyme and/or metabolite in it?
Integrative determination of metabolic pathways based on structure and systems information
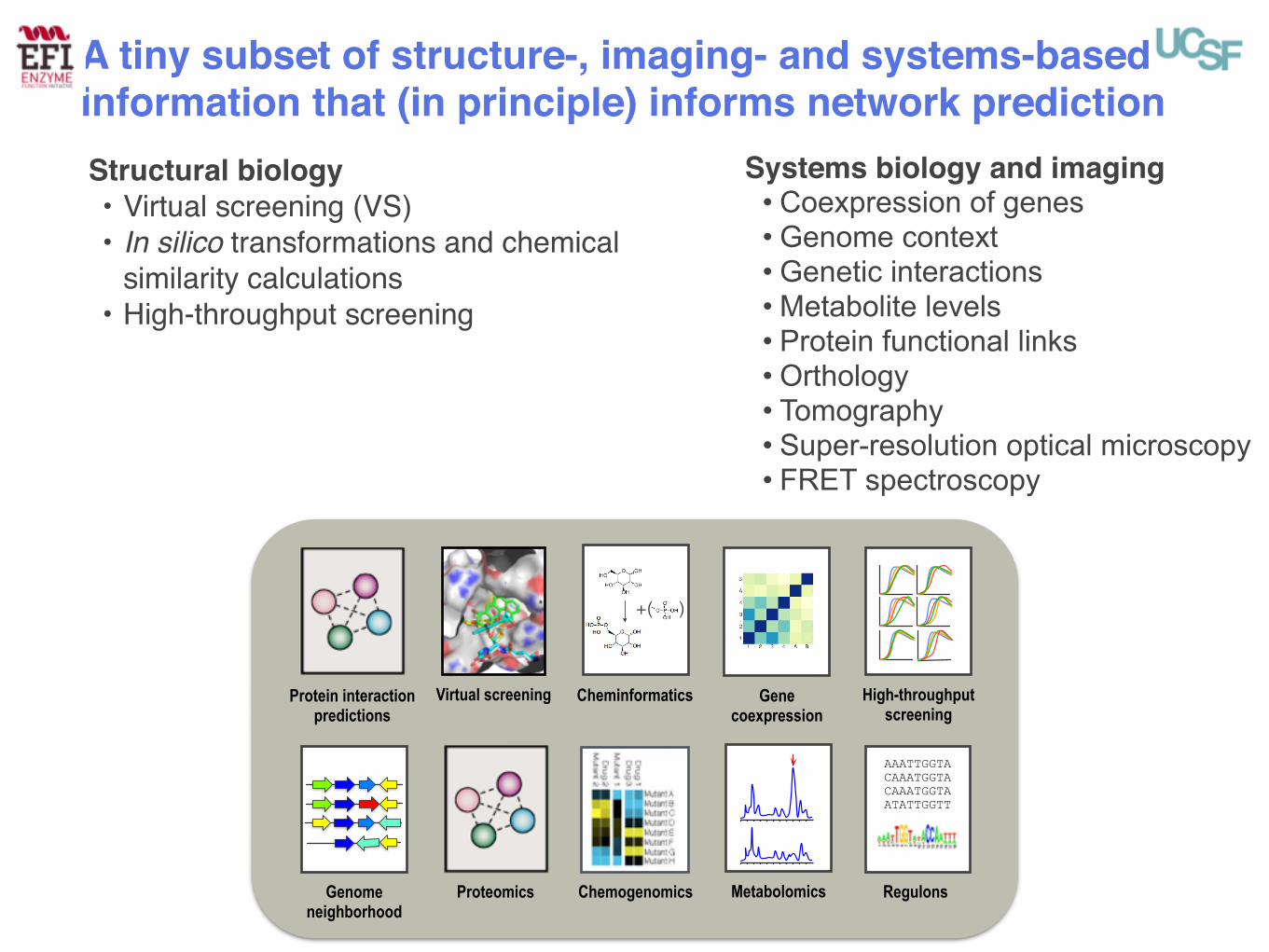
Proteomics Chemogenomics
Virtual screening Cheminformatics
Genome neighborhood
Protein interaction predictions
Gene coexpression
High-throughput screening
Metabolomics
AAATTGGTA CAAATGGTA CAAATGGTA ATATTGGTT
Regulons
+( )
Structural biology• Virtual screening (VS)• In silico transformations and chemical
similarity calculations• High-throughput screening
Systems biology and imaging• Coexpression of genes • Genome context • Genetic interactions • Metabolite levels • Protein functional links • Orthology • Tomography • Super-resolution optical microscopy • FRET spectroscopy
A tiny subset of structure-, imaging- and systems-based information that (in principle) informs network prediction

Components
Mindset: From integrative structure modeling towards integrative pathway mapping
Sampling and scoring
Model
Cross-linking
Restraints
Co-purification Bioinformatics, physics
Electron microscopy
NMRX-ray crystallography
Small angle X-ray scattering
Proteomics HDXMS
Sali et al. Nature, 2003
Alber et al. Nature, 2007
Robinson et al. Nature, 2007
Alber et al. Annual Reviews in Biochemistry, 2008
Russel et al. PLoS Biology, 2012
Ward et al. Science, 2013
IMP

Components
Mindset: From integrative structure modeling towards integrative pathway mapping
Sampling and scoring
Complex Topology
Model
Cross-linking
Restraints
Co-purification Bioinformatics, physics
Electron microscopy
NMRX-ray crystallography
Small angle X-ray scattering
Proteomics HDXMS
Sali et al. Nature, 2003
Alber et al. Nature, 2007
Robinson et al. Nature, 2007
Alber et al. Annual Reviews in Biochemistry, 2008
Russel et al. PLoS Biology, 2012
Ward et al. Science, 2013
IMP

Components
Mindset: From integrative structure modeling towards integrative pathway mapping
Sampling and scoring
Biological network
Model
Cross-linking
Restraints
Co-purificationDocking Bioinformatics, physics
Electron microscopy
NMRX-ray crystallography
Cheminformatics
Operon/genome context
IMPPotential

Components
Mindset: From integrative structure modeling towards integrative pathway mapping
Sampling and scoring
Metabolic Pathway
Model
Cross-linking
Restraints
Co-purificationDocking Bioinformatics, physics
Electron microscopy
NMRX-ray crystallography
Cheminformatics
Operon/genome context
IMPPotential
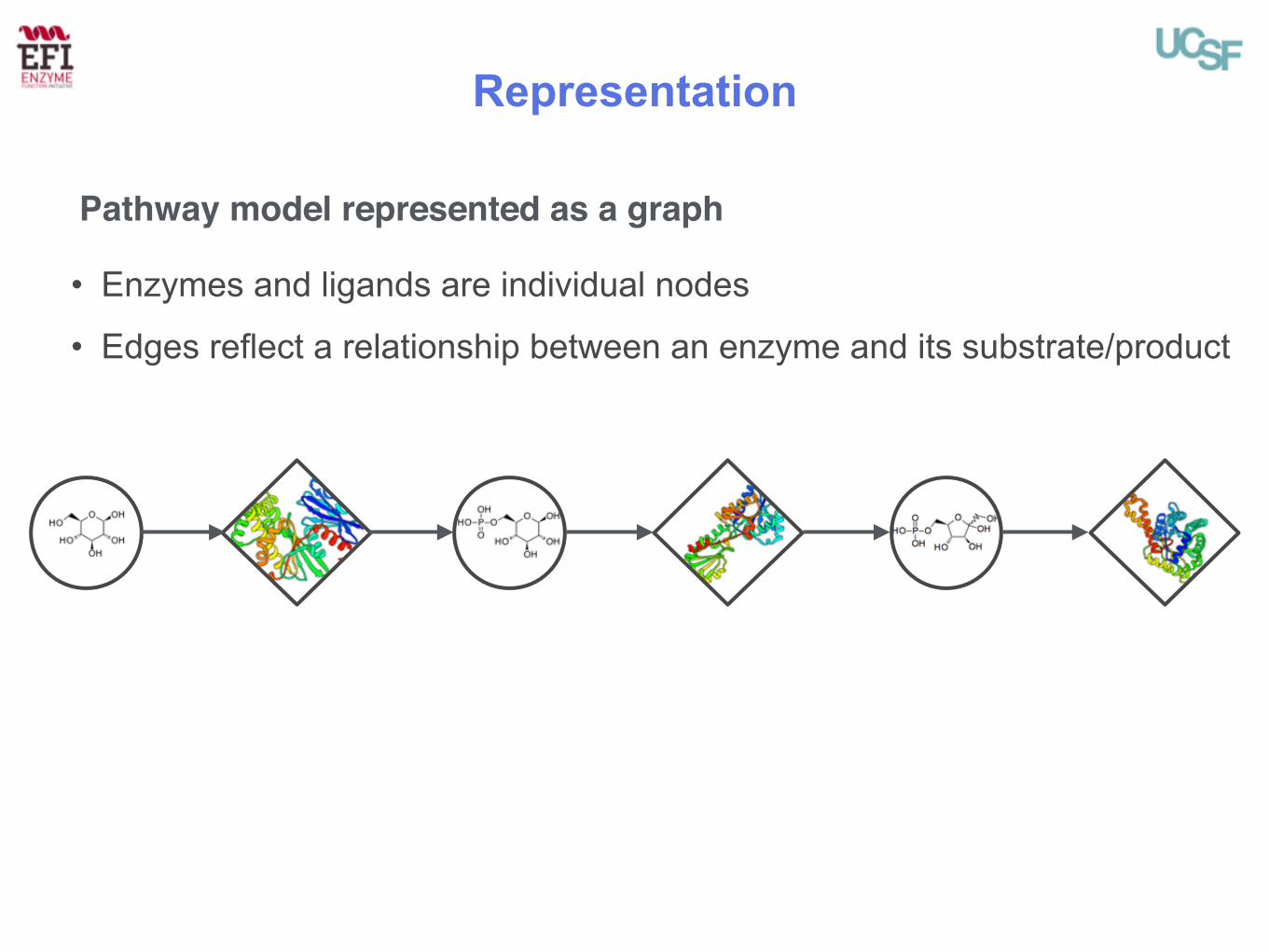
Representation
• Enzymes and ligands are individual nodes
• Edges reflect a relationship between an enzyme and its substrate/product
Pathway model represented as a graph

Scoring• Translation of information into restraints that quantify the degree of
consistency between a pathway model and the corresponding information • Scoring function is a sum of individual restraints to rank alternative models
Similarity ensemble approach (SEA) E-value
Chemical similarity between transformed substrate and product
Virtual screening (VS)docking score Similarity to screening hits

Sampling Monte Carlo with simulated annealing
1) Starting with a model, select a node to alter identity
2) Select a new node identity from all possible node identities & make change in model
3) If change improves score, accept the new model. Otherwise, accept the new model with a probability determined by the difference in scores.
4) Repeat until “convergence”
ΔS = Score(modeli+1) - Score(modeli)Prob. of acceptance = exp(-ΔS/T)

Example I: reconstructing glycolysis
1kinase
2isomerase
3kinase
4aldolase
5isomerase
6dehydrogenase
7kinase
8mutase
9enolase
10kinase
Here, we are treating glycolysis as a linear pathway

Four-stage overview Glycolysis
Docking data from: Chakrapani Kalyanaraman and Matthew P. Jacobson, Biochemistry 49, 4003, 2010. SEA calculations by Magdalena Korczynska, Henry Lin, and Brian Shoichet
Gatheringinformation
Analyzing models and information
Samplinggood models
Designing modelrepresentationand evaluation
• Assessing top models over 3.5 standard deviations above mean
• Monte Carlo with simulated annealing 5 M iterations per run & 400 runs
• 2,742 potential substrates & products • 10 glycolytic enzymes • Docking scores for ligand-enzyme pairs • SEA e-values between all enzymes • Chemical transformation scores for all
substrate-enzyme-product trios
• Fixed-length graph: 10 protein nodes and 11 ligand nodes
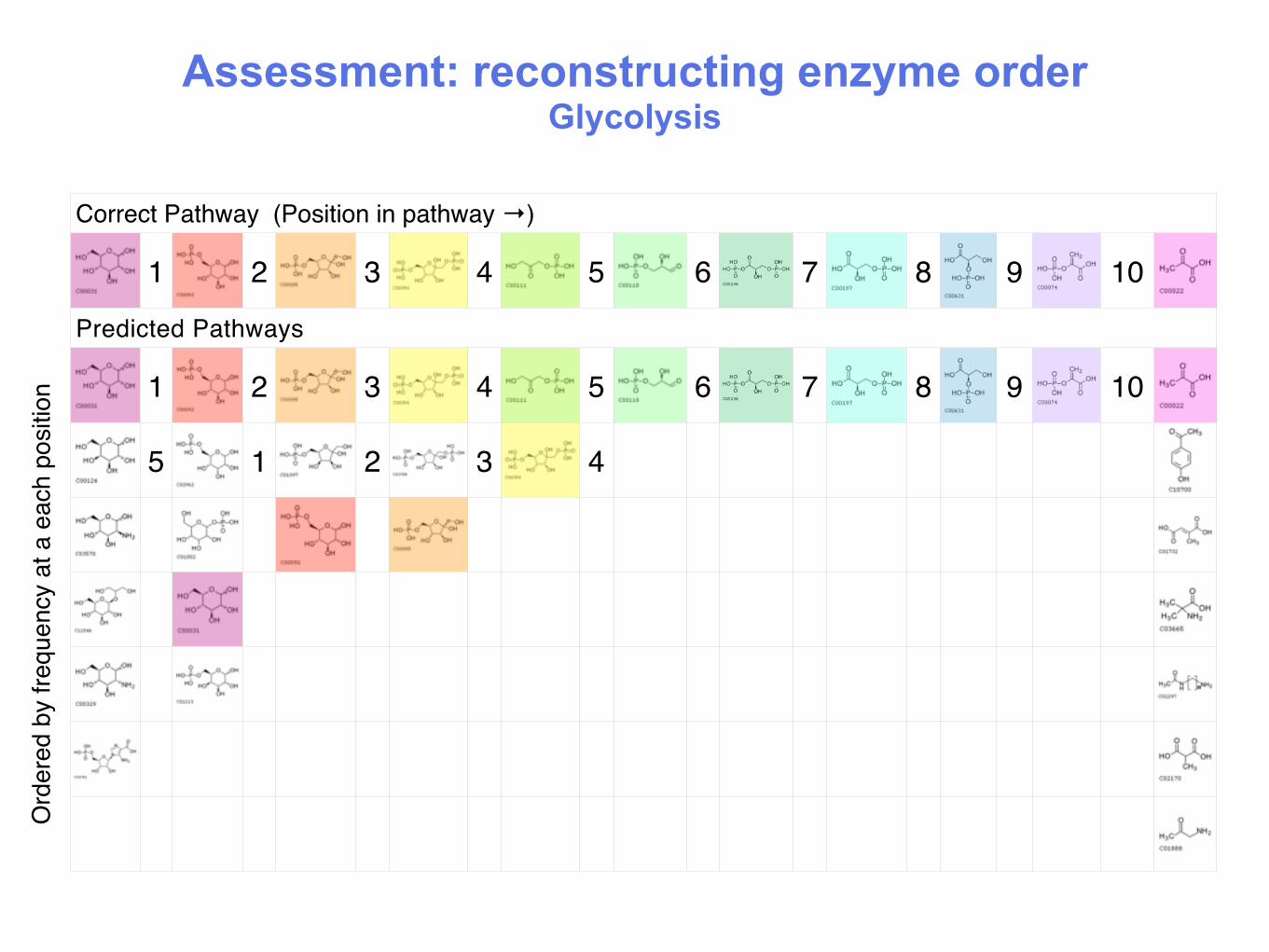
Assessment: reconstructing enzyme order Glycolysis
Ord
ered
by
frequ
ency
at a
eac
h po
sitio
n
Correct Pathway (Position in pathway →)
1 2 3 4 5 6 7 8 9 10
Predicted Pathways
1 2 3 4 5 6 7 8 9 10
5 1 2 3 4

Step in pathway
Rank by VS score alone
Rank using VS scores
Rank using chemical
transformations
Rank using VS, chemical
transformations, and SEA
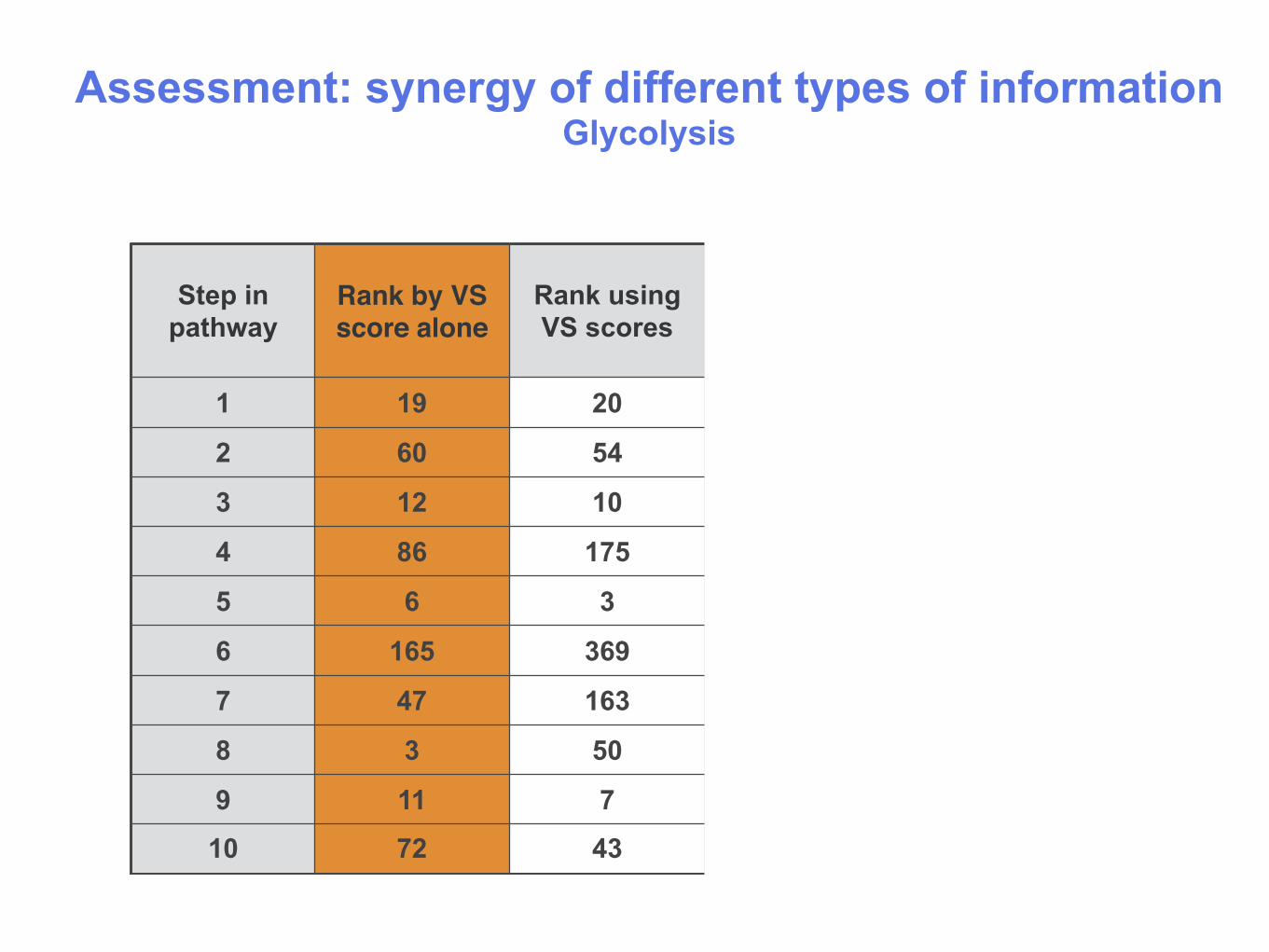
1 19 20 54 1
2 60 54 27 1
3 12 10 93 1
4 86 175 83 1
5 6 3 2 1
6 165 369 3 1
7 47 163 1 1
8 3 50 1 1
9 11 7 1 1
10 72 43 1 1
Assessment: synergy of different types of information Glycolysis
Step in pathway
Rank by VS score alone
Rank using VS scores
Rank using chemical
transformations
Rank using VS, chemical
transformations, and SEA
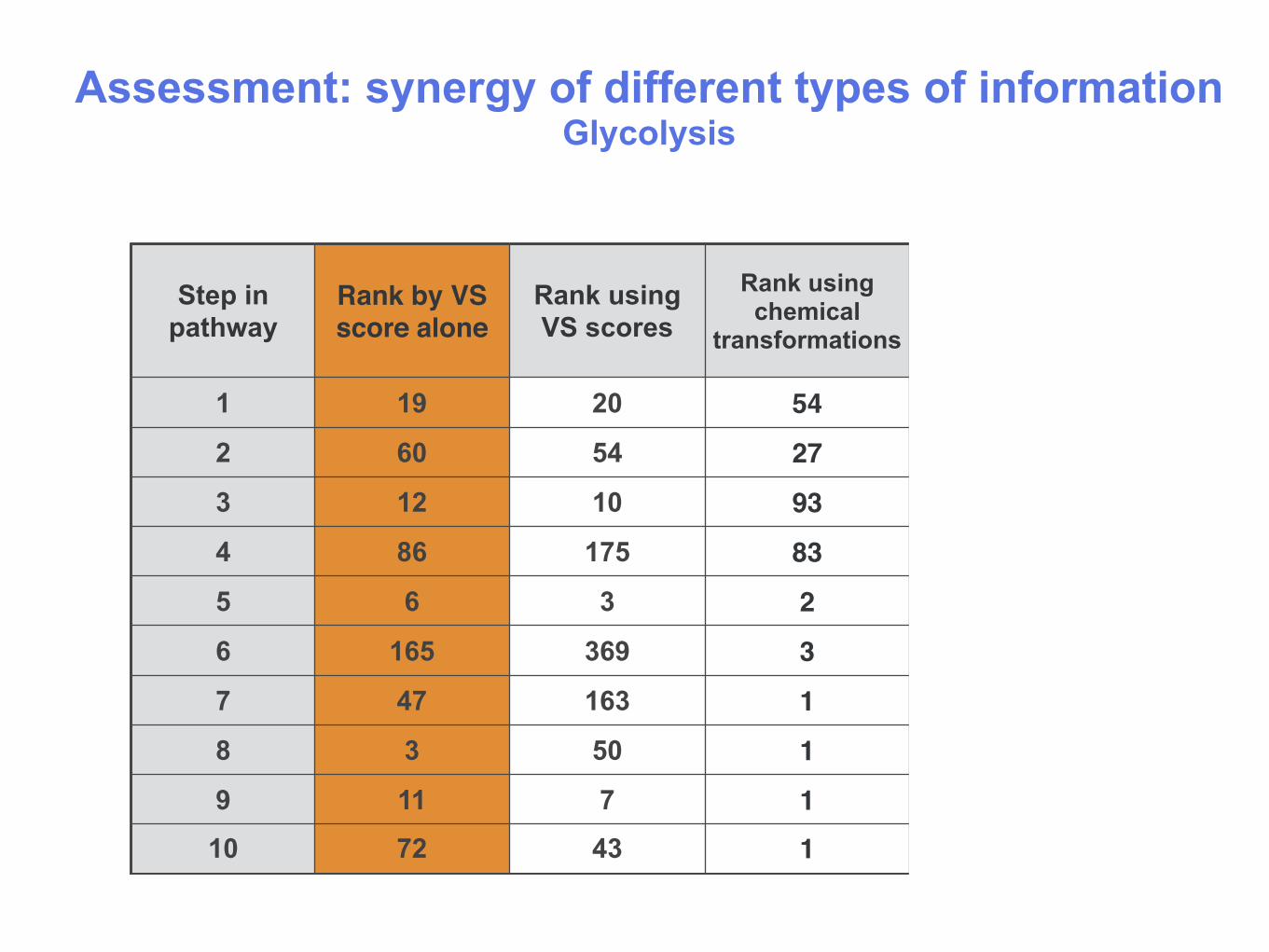
1 19 20 54 1
2 60 54 27 1
3 12 10 93 1
4 86 175 83 1
5 6 3 2 1
6 165 369 3 1
7 47 163 1 1
8 3 50 1 1
9 11 7 1 1
10 72 43 1 1
Assessment: synergy of different types of information Glycolysis
Step in pathway
Rank by VS score alone
Rank using VS scores
Rank using chemical
transformations
Rank using VS, chemical
transformations, and SEA
1 19 20 54 1
2 60 54 27 1
3 12 10 93 1
4 86 175 83 1
5 6 3 2 1
6 165 369 3 1
7 47 163 1 1
8 3 50 1 1
9 11 7 1 1
10 72 43 1 1
Assessment: synergy of different types of information Glycolysis

Step in pathway
Rank by VS score alone
Rank using VS scores
Rank using chemical
transformations
Rank using VS, chemical
transformations, and SEA
1 19 20 54 1
2 60 54 27 1
3 12 10 93 1
4 86 175 83 1
5 6 3 2 1
6 165 369 3 1
7 47 163 1 1
8 3 50 1 1
9 11 7 1 1
10 72 43 1 1
Assessment: synergy of different types of information Glycolysis
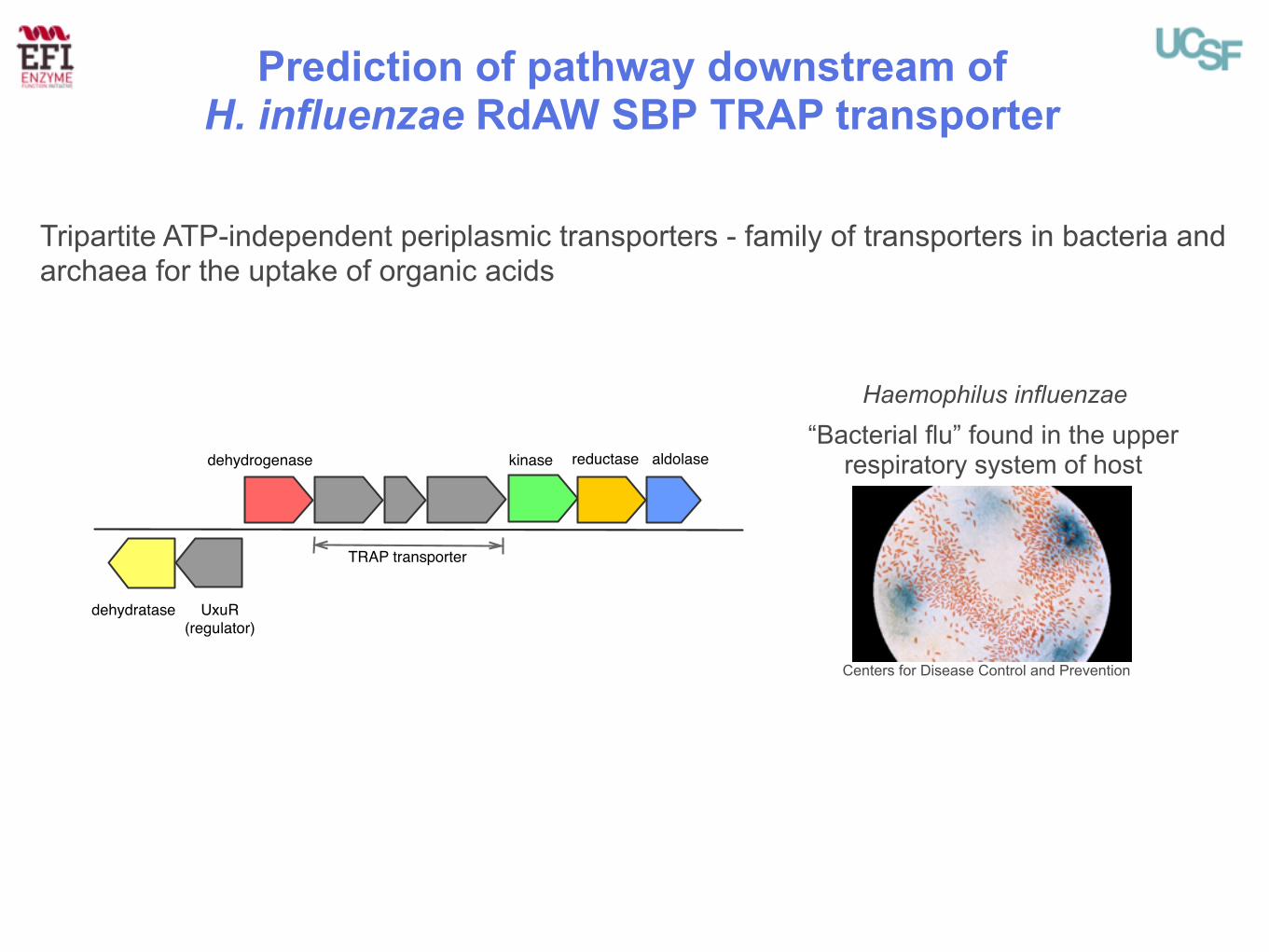
Prediction of pathway downstream of H. influenzae RdAW SBP TRAP transporter
TRAP transporter
dehydrogenase
UxuR (regulator)
dehydratase
aldolasekinase reductase
Haemophilus influenzae
Tripartite ATP-independent periplasmic transporters - family of transporters in bacteria and archaea for the uptake of organic acids
“Bacterial flu” found in the upper respiratory system of host
Centers for Disease Control and Prevention

Virtual screening Chemical transformations
Genome neighborhood
High-throughput screening
+( )Gathering information
Sampling good models
Designing model representation and evaluation
Pathway model
Analyzing models and information
Potential ligands Potential proteins
Similarity ensemble approach
Metabolic endpoints
…
6 protein-ligand pairs
5 enzymatic reactions
1 protein screened
1 endpoint 6 proteins6 proteins
Monte Carlo simulated annealing
Assessing sampling Assessing accuracy

Validated SPB TRAP pathway prediction
in vitro assays by Daniel Wichlecki, Gerlt lab Structure by Matthew Vetting et al., NYSGXRC

Metformin “network”Viollet, B., Guigas, B., Sanz Garcia, N., Leclerc, J., Foretz, M., & Andreelli, F. (2011). Cellular and molecular mechanisms of metformin: an overview. Clinical Science, 122(6), 253–270.

In ConclusionThe goal is a comprehensive description of the multitude of interactions between molecular entities, which in turn is a prerequisite for the discovery of general structural principles that underlie all cellular processes.
Sali, Earnest, Glaeser, Baumeister. From words to literature in structural proteomics. Nature 422, 216-225, 2003. Robinson, Sali, Baumeister. The molecular sociology of the cell. Nature 450, 974-982, 2007. Alber, Foerster, Korkin, Topf, Sali. Annual Reviews in Biochemistry 77, 11.1–11.35, 2008. D. Russel et al. “Putting the pieces together: integrative structure determination of macromolecular assemblies”. PLoS Biol., 2012. A. Ward, A. Sali A, I. Wilson. Integrative structural biology. Science 339, 913-915, 2013. Schneidman, Pellarin, Sali. Uncertainty in integrative structural modeling. Curr. Opin. Str. Biol., 96-104, 2014.
This goal will be achieved by a formal integration of experiment, physics, and statistical inference, spanning all relevant size and time scales.


















