
1 Clinical Trials. 2 This is part of drug registration but may be purely scientific: part of...
69
1 Clinical Trials
-
Upload
claud-booth -
Category
Documents
-
view
213 -
download
0
Transcript of 1 Clinical Trials. 2 This is part of drug registration but may be purely scientific: part of...
1
Clinical Trials

2
This is• part of drug registration
• but may be purely scientific: part of international multicentre trials for sponsors
In the legislation may be both
• general regulation of new biomedical research (e.g. new chirurgical techniques)
• and specific for clinical trials with investigational drugs

3
Clinical trials on human subjects
Very special ethical problems!
• research on human beings
• aiming new drug/indication development what means high profit
- new drugs
- rapid market access

4
In general, biomedical research on human beings
Ethical only if for new
• diagnostic methods,
• treatment opportunities,
• prevention and
• rehabilitation methods
different from those applied generally and/or with new substances/devices

Check what rules apply to clinical research in your country!
• It is highly possible that they are very similar to those I teach now…
• …for they are international, accepted also by WHO
5

6
Biomedical research on humans
Conditions differ, if the subjects are:
1.physically/mentally capable
2.physically/mentally incapable or restricted
3.vulnerable/dependent
Different regulation!

7
1. Research on physically-mentally capable subjects possible if
• The trial protocol has been authorised and/or ethically approved (see later)
• It is supported by previous studies (animal and human)
• The expected benefit/risk ratio acceptable• The purpose can not be achieved in other way
(e.g. by animal experiments)
• Informed consent of the trial subjects received

8
Informed consent, 1
It is the process by which a person (subject = patient/healthy volunteer) voluntarily confirms his/her willingness to participate in a particular clinical trial after having been informed of all aspects of the trial that are relevant to the subjects’ decision to participate

9
Informed consent, 2
It should be understood essentially as a documented process to ensure that the subject controls the decision on whether to participate in clinical research and to ensure that the subjects participation only occurs when the research is consistent with the subject’s values, interest and preferences

10
Informed consent, 2
• Informed consent is documented by means of a written, signed and personally dated Form (as a rule…)
• If the person is illiterate, the consent can be verbal if two impartial witnesses support it

The language used in the oral and written information about the trial, including the written informed consent Form, should be as non-technical, as practical and should be at a level of complexity, understandable to the subject and/or the subject’s legally acceptable representative, and the impartial witness where applicable
11
How to provide the information?

12
Necessary informationIn writing, after verbal and written information:• may be withdrawn any time• what is this trial, purpose, duration• what is going to happen with him/her, unpleasant (e.g.
awaken for drawing blood)
• what is the expected benefit• risks and compensation possibility• if no consent: what are the alternative treatment
possibilities• name of the competent investigator

13
Necessary information (continued)
• Confidental handling of his/her personal data
• Who will cover the trial costs? (Conflict of interest)
• The approximate number of trial subjects involved in the study

14
2. Research on physically/mentally incapable persons
• Similarly: The research protocol approved, previous studies gave
reason, benefit/risk acceptable, no other way (e.g. animal
experiments) for the study
• The trial is the person’s interest (some exceptions, see later)
• This research is impossible on capable persons• Consent of the legal representative (when only
restricted ability: also of the person himself/herself)

15
Research on physically/mentally incapable persons
Excemption from the “his/her own interest” requirement:
• the result - within calculable time - may serve the interest of similar persons example: vaccination trials!
• minimal risk• in some countries: higher authorisation
(e.g. Minister of Health)

16
Psychiatric incapable patients:
As a rule, further conditions, e.g. no suicide risk, the trial is not too long, no major depression...

17
3. “Vulnerable” persons
• Pregnant/breast feeding women may only be involved in the research if
own/child’s/similar others’ health interest
impossible on others
similar higher authorisation (Minister)

18
3. “Dependent” persons
• Persons dependent by service, financial, ethical reasons, as a rule, may not be involved
• In many countries: persons under military service never, arrested/prisoners only if his/her own health interest and the research can not be carried out on other persons

19
Excemptions from the consent of the involved persons
The medical intervention is very urgent and the new procedure, at a high probability, will serve the subjects’ health

20
Compensation paid for trial subjects
Principal rule (it may be different in human clinical trials with drugs): in many countries: if conducted properly but health damage: compensated by the trial site. For it, the trial site must have insurance (if such is available in the country, or abroad, if not: by the State)

21
Biomedical research involving human genes
If the purpose is not
• alteration of genes of descendants
• creation of a new species

Clinical trials on human subjects, as a rule, may not be commenced without
permission• In some countries, an independent
Ethics Committee should review the trial plan („protocol”) and approve it
• In other countries trials must be authorised (e.g. by Ministry of Health, or the Drug Agency if such exists)
• In other countries both are needed• The situation in your country?
22

23
Ethical (review) Committee
• Independent
• Not only physicians, also „lay” (from health-sciences point of view) persons: lawyer, clergyman, ethicist, psychologist, possibly patient organisation representative
• Appeal is possible against its opinion/approval

The Ethics Committee…
• Checks and approves the ethics of the trial (see points of view discussed before, e.g. circumstances of the informed consent)
• If no authority review: also the scientific part (is the trial suitable, according to its protocol, for its intended purpose?)
24

25
Ethical review
• Protocol scientifically sound?• Adequate treatment provided? homeopathy? • Benefit/risk acceptable?• Investigator and the trial site suitable?• Information sheet and consent Form• Insurance coverage acceptable?• Placebo application reasoned (if applicable)

26
(continued)
• compensation of tial subjects• remuneration of trial subjects (if
applicable) and of the investigator• recruitment methods• reason of involvement of pregnant,
prisoner, mentally incapable subjects a • content of the Investigators’ Brochure • Personal data protection

27
(continued)
Ethical Committee may request further data (in EU only once)
Then written opinion
• approved
• modification
• rejected

28
Clinical trials on human subjects with (investigational)
products
• As a rule (e.g. in the EU) maximum time-frame for approval/rejection
• Ethical Committee approval plus Drug Regulatory Authority permission (or notification)

29
Definitions, 1
• Investigational product: for legal reasons not called drug, active substance or placebo in dosage-form
• Clinical trial with investigational products to study its efficacy, side-effects, tolerability, pharmacodymanics and -kinetics (ADME), bioavailability, on the basis of a pre-approved trial protocol

30
Definitions, 2• New therapeutic indication: the product is
registered but not for this indication• Non-interventional trial: the product is
used as registered, there is no trial protocol, the product not administered for research purpose, the patient is treated normally, but the data are evaluated using epidemiological (statistical) methods

31
Definitions, 3
• Ethical Committee not only physicians! for ethical approval of the trial protocol, may be Central/Regional and local (hospital)
• Informed consent the patient is informed verbally and in writing, he/she (legal representative) signs the informed consent form

32
Definitions, 4
• Clinical trial phases Phase I, II, III, bioequivalence, IV
• Phase I CT pharmacokinetics and tolerability of the product, cca. 10 healthy volunteer (unless the product is highly toxic). For:
THE HUMAN BEING IS NOT A BIG RAT!

33
Definitions, 5
• Phase II CT on a few, around 100 patients: same therapeutic effects observed on animals?
• Phase III CT normally a few thousand patients: efficacy, frequent side-effects, special groups elderly, children...
THE CHILD IS NOT A SMALL ADOULT!

34
Definitions, 6
• Bioequivalence CT comparative PK (blood levels)
• Phase IV CT with registered drug, e.g. rare side-effects. More intervention (examinations, blood tests, etc.) than in normal treatment

35
Definitions, 7
• Multicentre trials for there are not enough patients in one single trial site. Coordinating investigator
• Controlled — open CTs comparison with another drug product or placebo
• Randomised CT subjects enlisted in the treated with the investigational drug (called verum) or in the control arm in a random order

36
Open clinical trialAll patients treated with the investigational
medicinal product less informative than a controlled trial
37
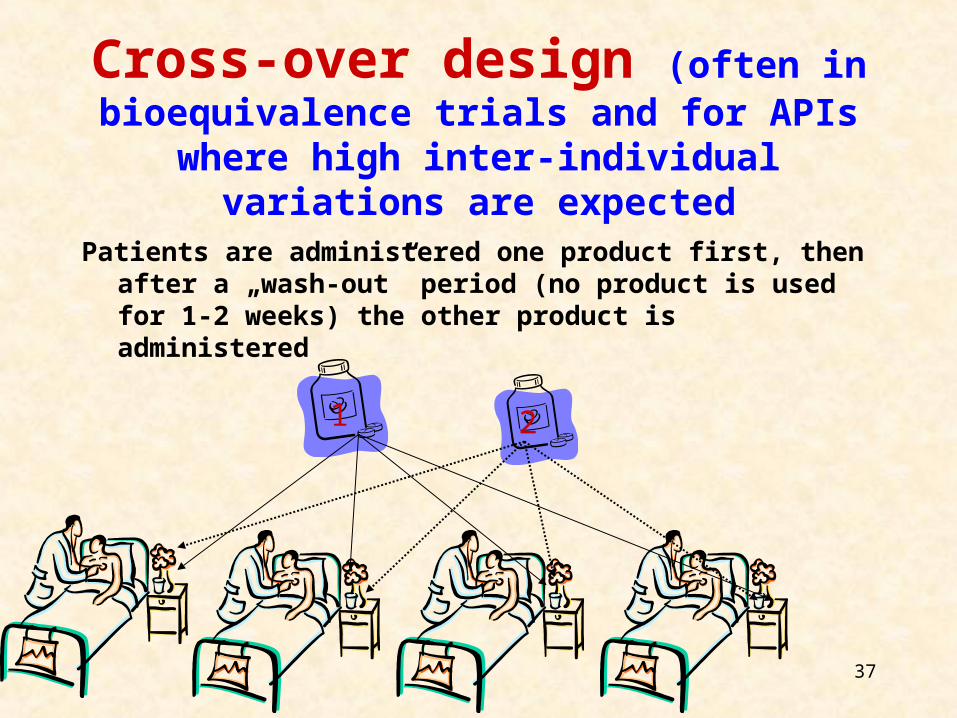
Cross-over design (often in bioequivalence trials and for APIs where
high inter-individual variations are expected
Patients are administered one product first, then after a „wash-out” period (no product is used for 1-2 weeks) the other product is administered
1 2

38
Controlled trial
The first group (=arm), verum receives the new product while the other (control) treatment arm the comparator product or placebo
1 2

39
Definitions, 8
• Double blind - single blind. Blinding single: the patient, double: netiher the patient nor the doctor knows whether the patient is in the verum or in the control arm - coded products! - careful explanation in ophtalmologic trials...
• Sponsor who initiates, directs and/or finances the CT

40
Definitions, 9• Side-effect or adverse effect (ADR) on
the basis of the previous studies, not expected/not desired effects
• Serious ADRs• Adverse event (ADE) happens to the
patient during the CT, not necessarily caused by the investigational product (e.g. abnormal labour result, patient falling off the bed…)

41
Definitions, 10
• Standard Operation Procedure SOP description of an examination e.g. measurement of body temperature
• Case Report Form CRF printed Form, to write the examination/treatment data in
• Investigator’s Brochure summary of previous preclinical and/or clinical data

42
Definitions, 11
• Standard Operation Procedure SOP description of an examination e.g. measurement of body temperature
• Case Report Form CRF printed Form, to write the examination/treatment data in
• Investigator’s Brochure summary of previous preclinical and/or clinical data

43
Definitions, 12
• Inspector on-the-spot inspection on behalf of the DRA, may look into personal patient data, thus, as a rule, doctor or pharmacist
• Monitor similar, auditing the CT on behalf of the Sponsor

44
Definitions, 13
• Protocol purpose of the CT, conduct, modification, statistical methods. Compiled and approved previously, together with the possible modifications
• Participant a subject (patient or healthy volunteer) involved into the CT

45
General rules for the CT• It may not be a substitution of the
treatment
• If persons of reproductive age: emphasis whether it may impair reproductive functions?
• The possibility of causing health damages must be minimised
• Health status of subject controlled continuously

46
Involving subjects
• General rule: those who are present at the trial site (hospital)
• Recruitment (e.g. by media) possible in certain countries. In another countries this possible only for healthy volunteers, otherwise only doctors may be informed on CT possibilities

47
Information to subjects
• Verbally, in writing (if not illiterate) in his/her language, in an understandable way
• In the document: is/is not physically/mentally capable
• The subject may receive a copy from the information sheet, one copy goes to the CT documentation

48
This information, as a minimum:
• Identifying data of the CT• This is a CT: its time-period, how many
persons involved, conduct type and frequency of examinations/interventions,
• which are the other “normal” treatment possibilities, possible consequences of termination of the treatment started previously (if applicable) ...

49
(continued)
• Foreseeable risks (pointing out that may be unforeseeable ones, too)
• What is the benefit (or: it is not expected for the concrete subject)
• Possibility of random assignment for the verum and control arms

50
(continued)
• in case of any health injury: the treatment, compensation, the name of the insurance company contracted
• financial compensation (if applicable)
• the consent may be withdrawn any time, without giving any reason
• personal data protection rules

51
(continued)
• Denomination of the local/hospital Ethical Committee, name of its Chairperson (if such does exist)
• what is placebo, he/she may be assigned to it (if applicable)
• short description of the expected effects of the investigational drug product

52
The (informed) consent Form
• Identifying data of the CT• Name of the CT site (hospital)• Name of the investigator (or that of
giving the information), position• Trial subject’s data• In case of (restricted) physical/mental
ability: the same of the legal representative

53
(continued)
• The consent has been given after information, without any influence, knowing that it can be withdrawn any time
• date of the signature• signature of the investigator/the
person giving the information• signature of the consenting person

54
Witnesses?
• Only when the person, giving the consent (or his/her legal representative) is illiterate
• then two impartial witnesses

55
Further informed consent rules
• If new important information: start the whole process again
• Restricted capability: also this person, as far as his/her ability permits it
• Informed consent of minors: person with pedagogic experience present

56
(continued)
• If the subject rejects his/her involvement, this must be accepted even if he/she is mentally incapable
• The same when rejected in a mentally capable state but later becomes incapable
• If the opposite: must be informed accordingly

57
International dispute
Should the control group’s treatment be covered by the State (insurance) Fund (as “ordinary” patients or should their treatment financed by the Sponsor?

58
Manufacture of the investigational product
• GMP special chapter. Manufacture: as a rule, the first time and at a small scale. Labelling special (control, placebo! Coding)
• Phase IV: naturally marketed product, but labelling as: “Investigational sample, not for marketing”

59
Adverse Drug Reaction (ADR) reporting
• Serious ADR or ADE: investigator reports to Sponsor and Ethics Committee immediately
• other ADR/ADE: according to the protocol
• The ADE (not necessarily caused by the drug) is death: <15 days (continued)

60
(continued)
• Any ADR causing death or serious health injury, even if suspected: investigator reports to Ethics C’ttee, the competent authority and other trial sites <7 days then within +8 days to Sponsor all info needed for the decision whether the trial can be followed

61
(followed)
• The sponsor records all serious (suspected) ADR/ADE occurred anywhere (multicentre trials in many countries!)
• If needed, the competent authority may have access to this database
• Report to Competent authorities and Ethics C’ttees annually

Good Clinical Practice (GCP)
An internationally accepted quality assurance system for planning. Conducting and reporting clinical trials on human beings
62

63
Placebo in CTs
Special prerequisites • there is no evidence-based treat-ment of the
illness/symptom or• there is one, but toxic and not well tolerated,• and the postponement of real treatment does
not represent a significant risk to the patient• it is only additional to the best available
treatment (“add on”)

64
Good Clinical Practice [for research with investigational
products]
International guideline
GCP

65
GCP Principles
• Well-being, health and integrity of trial subjects supersedes expected scientific results
• The expected benefit/risk rate was already characterised by preclinical (animal) studies, and is is known by the investigator and, also to some extent, by the trial subjects

66
Main chapters of the GCP
• The Ethical Committee
• The investigator
• Informed consent of the trial subjects
• The Sponsor
• The CT protocol

67
Inspection of CTs
State GCP Inspectorate (if any):
Manufacture of the investigational products
At the trial site:• data handling and documentation• compliance with GCP• handling of the investigational product

Exam topic!
68

Clinical trials regulation• Biomedical research on human beings: when is it ethical?
• Outline the different regulation for „normal”, physically/mentally incapable and vulnerable/dependent persons, including their definitions and special conditions
• Prerequisites for a biomedical research on human beings
• What is informed consent?
• How to inform patients?
• The Ethics Committee and its activities. Content of its review
• Clinical trials with investigational medicinal products. „Double permission”
• Certain important definitions
• Clinical trial phases
• General rules, compensation
• ADR and ADE, their reporting
• Clinical trial inspection. What is GCP?
69


















