
α-Thrombin mediated PI 3-Kinase activation through release ... · α-Thrombin mediated PI 3-Kinase...
50
α-Thrombin mediated PI 3-Kinase activation through release of Gβγ dimers from Gα q and Gα i2 . Reema Goel ∗ , Polly J. Phillips-Mason ∗† , Alice Gardner ∗± , Daniel M. Raben § and Joseph J. Baldassare ∗ ¶ From the ∗ Department of Pharmacological and Physiological Sciences, St Louis University Medical School, St. Louis, Missouri 63104 and the § Department of Physiology, The Johns Hopkins University School of Medicine, Baltimore, Maryland 21205. ¶To whom correspondence should be addressed: Department of Pharmacological and Physiological Sciences, St Louis University Medical School, 1402 South Grand Blvd., St. Louis, Missouri 63104; Tel.: 314-577-8543; Fax: 314-577-8233; E-mail: [email protected] Running title: PI 3-Kinase pathway requires Gα q and Gα i2 associated Gβγ dimers. 1 by guest on December 25, 2019 http://www.jbc.org/ Downloaded from
Transcript of α-Thrombin mediated PI 3-Kinase activation through release ... · α-Thrombin mediated PI 3-Kinase...

α-Thrombin mediated PI 3-Kinase activation through release of Gβγ dimers from Gαq and
Gαi2.
Reema Goel∗, Polly J. Phillips-Mason∗†, Alice Gardner∗±, Daniel M. Raben§ and Joseph J.
Baldassare∗¶
From the ∗Department of Pharmacological and Physiological Sciences, St Louis University
Medical School, St. Louis, Missouri 63104 and the §Department of Physiology, The Johns
Hopkins University School of Medicine, Baltimore, Maryland 21205.
¶To whom correspondence should be addressed: Department of Pharmacological and
Physiological Sciences, St Louis University Medical School, 1402 South Grand Blvd., St. Louis,
Missouri 63104; Tel.: 314-577-8543; Fax: 314-577-8233; E-mail: [email protected]
Running title: PI 3-Kinase pathway requires Gαq and Gαi2 associated Gβγ dimers.
1
by guest on Decem
ber 25, 2019http://w
ww
.jbc.org/D
ownloaded from

SUMMARY
Chinese hamster embryonic fibroblasts (IIC9 cells) express the Gα subunits Gαs, Gαi2,
Gαi3, Gαo, Gαq/11 and Gα13. Consistent with reports in other cell types, α-thrombin stimulates a
subset of the expressed G proteins in IIC9 cells, namely Gi2, G13 and Gq as measured by an in
vitro membrane [35S]GTPγS binding assay. Using specific Gα peptides, which block coupling of
G-protein receptors to selective G proteins, as well as dominant negative xanthine nucleotide
binding Gα mutants, we show that activation of the Phosphatidylinositol 3-Kinase /Akt pathway
is dependent on Gq and Gi2. To examine the role of the two G proteins, we examined the events
upstream of PI 3-Kinase. The activation of PI 3-kinase/Akt pathway by α-thrombin in IIC9 cells
is blocked by the expression of dominant negative Ras and β-arrestin1 (Phillips-Mason, P. J.,
Raben, D. M., and Baldassare, J. J. (2000) JBC 275, 18046 and Goel, R., Phillips-Mason, P. J.,
Raben, D. M., and Baldassare, J. J. (2002) JBC 277, 18640), indicating a role for Ras and β-
arrestin1. Interestingly, inhibition of Gi2 and Gq activation blocks Ras activation and β-arrestin1
membrane translocation, respectively. Furthermore, expression of the Gβγ sequestrant, α-
transducin, inhibits both Ras activation and membrane translocation of β-arrestin1, suggesting
that Gβγ dimers from Gαi2 and Gαq activate different effectors to coordinately regulate the PI 3-
Kinase/Akt pathway.
2
by guest on Decem
ber 25, 2019http://w
ww
.jbc.org/D
ownloaded from

INTRODUCTION
α-Thrombin is a potent mitogen for Chinese hamster embryonic fibroblasts (IIC9) (1).
The α-thrombin receptor is a member of the 7 membrane spanning family of G-protein coupled
receptors (GPCRs) known as the Proteinase Activated Receptors (PARs) (2). Upon activation,
GPCRs catalyze the exchange of GTP for GDP on the Gα subunit of specific G protein
trimers, promoting the dissociation of Gα-GTP from the Gβγ subunits, which remain tightly
associated (3). Both the Gα-GTP and the Gβγ dimers stimulate effectors including
phospholipase C (4), Ras (5) and PI 3-Kinases (6,7).
Twenty distinct Gα subunits have been cloned which can be divided into four families
based on sequence homology: Gs, Gi, Gq and G12 (8). Members of the Gi family can be
identified by their sensitivity to ADP-ribosylation by pertussis toxin (PTX), which uncouples
the G protein from the receptor (9). In addition to the twenty Gα subunits, five Gβ subunits
and twelve Gγ subunits have been identified (10). The downstream events associated with a
particular GPCR depend on the heterotrimeric G proteins that associate with the receptor.
Several G protein coupled receptors, including α-thrombin, are known to activate multiple G
proteins and these G proteins can activate different signaling pathways (11-20). IIC9 cells
express Gs, Gi2, Gi3, Go, Gq/11 and G13 (21). Pertussis toxin-sensitive G proteins are involved
in α-thrombin-induced Ras activation (22), and Gq family members mediate the activation of
PLC-β1 (4) and PKC (22).
We have previously found that in IIC9 cells α-thrombin induces PI 3-Kinase and Akt
activities that are dependent on Ras and β-arrestin1 (23,24). At present the Gα subunits that
mediate activation of PI 3-Kinase and Akt by α-thrombin are unknown. This paper identifies
the specific Gα subunits by using transient expression of specific Gα peptides, which block
coupling of GPCRs with selective G proteins, as well as dominant negative xanthine nucleotide
3
by guest on Decem
ber 25, 2019http://w
ww
.jbc.org/D
ownloaded from

binding Gα mutants. We show that α-thrombin stimulates Gq, G13 and Gi2 in IIC9 cells.
Interestingly, the PI 3-Kinase and Akt stimulation are sensitive to uncoupling of both Gq and
Gi2. Efforts to delineate the individual role of Gq and Gi2 in this common pathway show that
Ras activation and the translocation of β-arrestin1 to membranes are mediated by Gβγ dimers
released from Gαi2 and Gαq respectively. These data indicate that α-thrombin induces PI 3-
Kinase and Akt stimulation via the Gβγ dimers from Gαq acting cooperatively with the Gβγ
dimers from Gαi2, and that PI 3-Kinase activation is a point of convergence in the action of the
two G proteins.
4
by guest on Decem
ber 25, 2019http://w
ww
.jbc.org/D
ownloaded from

MATERIALS AND METHODS
Cell Culture and Reagents- IIC9 cells, a clonal population of Chinese hamster embryo
fibroblasts were maintained in Dulbeco’s modified Eagle’s Medium (DMEM) containing 4.5
g/L glucose and 2mM L-glutamine (BioWhittaker, Walkersville, MD) supplemented with 5%
(v/v) fetal calf serum. Subconfluent IIC9 cells (80%) were growth-arrested by washing once
with alpha-MEM (Life Technologies), containing 2mM L-glutamine (BioWhittaker) followed
by 48-hour incubation in the same medium. Human α-thrombin isolated from plasma (Sigma,
St. Louis, MO) was used at 1 NIH unit/ml in all experiments and PTX (BioMol, Plymouth
Meeting, PA) was used at 100 ng/ml.
Transient Transfection- The cDNA encoding pcDNA3.1 (Invitrogen), β-arrestin1 (a kind gift
from Marc Caron), α-transducin (25), MAS-GRK3-ct (a kind gift from Stephen R. Ikeda),
GoαC351G, Gi2αC352G, and Gi3αC351G (generated), carboxyl terminus Gα peptides (cue
Biotech, Chicago, IL) or xanthine nucleotide binding Gα mutants (Guthrie Research Institute,
Sayre, PA) were transfected into subconfluent (60-80%) IIC9 cells using LiptofectamineTM
(Gibco BRL, Gaithersburg, MD) as previously described (23). The following day, cells were
growth-arrested by washing once with alpha-MEM followed by 48-hour incubation in the same
medium prior to agonist stimulation. Transient transfection using LiptofectamineTM resulted in
80-90% expression efficiency as visualized by co-transfection of GFP.
Western Blot Analysis- Growth-arrested IIC9 cells were incubated in the absence or presence of
indicated mitogen for times indicated in the figure legends. At the indicated times, cell lysates
were prepared as previously described (23). Protein lysates (10-25 µg) were resolved by SDS-
PAGE and transferred to a polyvinylidene diflouride membrane (Millipore, Boston, MA).
Membranes were probed with polyclonal antibodies to Akt (Santa Cruz Biotechnology),
5
by guest on Decem
ber 25, 2019http://w
ww
.jbc.org/D
ownloaded from

phospho-AktSer473 (Cell Signaling), phospho-ERK (Cell Signaling), ERK (Santa Cruz
Biotechnology), β-arrestin1 (Transduction Laboratories) and PI 3-Kinase p85 antibody (Upstate
Biotechnology, NY). Immunoreactive bands were visualized by enhanced chemiluminescence
(ECL) (Amersham Life Sciences, Arlington Heights, IL) as recommended by the manufacturer.
Preparation of IIC9 Membranes and β-Arrestin1 Translocation Assay- Plasma membranes from
IIC9 cells were prepared as previously described (23). The protein amounts of β-arrestin1 were
determined by Western Blot Analysis and quantified by using Molecular Dynamics
Phosphoimager.
Generation of PTX-Resistant Mutants- PTX-resistant Goα, Gi2α and Gi3α mutants were
generated using PCR to amplify these inserts from their current pcDNA vectors with the
addition of both a 5’ and 3’ restriction site and the C-terminal C→G mutation. For Goα a 5’
Hind III site and a 3’ Xba I site were introduced in addition to the C→G mutation using the
following primers (5’→3’): TAT AAA GCT TGG CCA CCA TGG GAT GTA CTC and TAT
ATC TAG ATC AGT ACA AGC CTC CGC CCC GGA GAT TGT T. For Gi2α a 5’ Hind III
site and a 3” Xho I site were introduced in addition to the C→G mutation using the following
primers (5’→3’): TAT AAA GCT TGG CAG GAT GGG CTG CA and TAT ACT CGA GTC
AGA AGA GGC CTC CGT CCT TCA GGT TGT TCT TG. For Gi3α a 5’ Hind III site and a 3”
Xho I site were introduced in addition to the C→G mutation using the following primers
(5’→3’): TAT AAA GCT TGG CCG CCG TCA TGG GCT GC and TAT ACT CGA GCC
TCT CAG TAA AGC CCA CCT TCC. Restriction sites are underlined. PCR was carried out in
the presence of 4 mM MgSO4 using the Vent enzyme (Promega) and the following cycling
conditions: 95°C, 5 minutes; 95°C, 1 minute, 55°C, 1 minute, 72°C, 1.5 minutes, 30 cycles. PCR
6
by guest on Decem
ber 25, 2019http://w
ww
.jbc.org/D
ownloaded from

products were restricted with the appropriate enzymes and subcloned into pcDNA3 (Invitrogen).
Mutants were screened by restriction enzyme analysis and mutations confirmed by sequencing
using a Perkin Elmer automated sequencer.
ADP Ribosylation Assay- IIC9 cells were pretreated with pertussis toxin at 100 ng/ml. IIC9
membranes were prepared as described as mentioned above and resuspended in 30 µl of 50 mM
Tris-HCl, pH 7.5, 1 mM EDTA and 1 mM MgCl2 to each sample 50 µl 2x reaction buffer (2mM
ATP, 100 mM Tris-HCl, pH 7.5, 20 mM thymidine, 40 mM arginine, 0.4 mg/ml BSA, 200 mM
KPO4, pH 7.5, 10 mM ADP-ribose, 20 mM MgCl2, 2 mM EDTA and 200 µM GTP) and 4 µl
activated pertussis toxin (pertussis toxin was activated by incubating 20 µl pertussis toxin
[1mg/ml] with 42 mM DTT, 20 mM Tris-HCl, pH 7.5 and was incubated at 30 °C for 30
minutes ). Finally, 10 µl of 5 µM NAD containing 20 µCi [32P]NAD was added to each sample
and was incubated at 30 °C for 30 minutes. The reaction was stopped by adding 100 µl of NAD
wash solution (5 mM NAD, 50 mM Tris-HCl, pH 7.5, 2.5 mM EDTA) to each sample.
Membranes were pelleted by centrifugation at 14,000 x g at 4°C for 5 minutes, washed twice
with 200 µl of NAD wash solution, resuspended in Laemmli sample buffer and boiled for 5
minutes. Proteins were separated by SDS-PAGE gel containing 6M urea and quantified using a
Molecular Dynamics Phosphoimager.
Ras/Rho Activity Assays- Growth-arrested IIC9 cells or transfected cells were incubated in the
presence or absence of 1 unit/ml α-thrombin for 5 minutes. After 5 minutes, the cells were
washed two times with 4oC PBS and harvested by scraping into 500 µl lysis buffer (50 mM Tris-
HCl, pH 7.5, 20 mM MgCl2, 150 mM NaCl, 1% Triton X-100, 2 mM p-nitrophenylphosphate,
10 µg/ml pepstatin, 10 µg/ml aprotinin and 10 µg/ml leupeptin), and the cell extracts centrifuged
7
by guest on Decem
ber 25, 2019http://w
ww
.jbc.org/D
ownloaded from

for 2 minutes at 4o C. The cell extract was added to glutathione beads complexed with GST-
RBD (fusion protein containing the Ras-binding domain of Raf-1 for Ras activity and fusion
protein containing the RhoA binding domain of Rhotekin for Rho activity) for 1 hour at 4o C.
Samples were washed three times with 500 µl of ice cold lysis buffer, analyzed on 15% SDS-
PAGE gel and transferred to PVDF. The membranes were probed with a 1/1000 dilution of a
pan Ras antibody (Santa Cruz) or RhoA monoclonal antibody (Santa Cruz). The amount of
activated Ras or Rho (complexed with GTP) was visualized by ECL detection.
Phosphatidylinositol 3-Kinase Assay- PI 3-Kinase activity determined as previously described
(23).
Assay of [35S]GTPγS Binding- Membranes (approximately 40µg protein) from serum arrested
IIC9 cells, were resuspended in 50µl of 50 mM Tris-HCl (pH 7.6), 2 mM EDTA, 100 mM
NaCl, 1 mM MgCl2, 1 µM GDP, 10 µg/ml pepstatin, 10 µg/ml aprotinin, 10 µg/ml leupeptin
and 50 nM [35S]GTPγS (2000 Ci/mmol), were incubated in the presence or absence of 1 unit/ml
α-thrombin at 37oC. After 5 minutes the reaction was terminated by the addition of 500µl of 50
mM Tris/HCl (pH 7.6), 150 mM NaCl, 20 mM MgCl2, 100 µM GDP, 100 µM GTP, 1%
Nonidet P-40, 10µg/ml pepstatin, 10 µg/ml aprotinin and 10 µg/ml leupeptin. The extracts were
incubated with 3µl of preimmune serum and 200µl of Pansorbin cells (Calbiochem) and
centrifuged after 30 minutes to remove nonspecifically bound proteins. Extracts were incubated
with antibody directed against a specific Gα subunit and incubated for 1 hour at 4oC. The
immune complex is then incubated with 50µl of a 50% protein G agarose suspension, the
complexes collected and washed three times in assay buffer plus 1% Nonidet P-40 and twice in
assay buffer without detergent and the presence of Gα subunits was analyzed by Western blot
8
by guest on Decem
ber 25, 2019http://w
ww
.jbc.org/D
ownloaded from

analysis using Gα specific antibodies. [35S ]GTPγS binding in the immunoprecipitates were
quantified by scintillation counting.
Confocal microscopy- Growth-arrested or transfected IIC9 cells grown on chamber slides
(Nalgeen®Labware, Rochester, NY) were incubated in the absence or presence of 1 unit/ml α-
thrombin for indicated time points after pre-incubation in the absence or presence of 100 ng/ml
PTX for 6 hours. Subsequent to activation, the cells were fixed in a 3.7% Formalin (Sigma)
solution for 10 minutes at room temperature followed by 6 minute incubation in ice cold
methanol at -20°C. The cells were washed in PBS and then blocked in 1 ml of blocking buffer
(0.8 g Fatty Acid Free BSA (Sigma) in 100µl PBS) for 2 hours at room temperature. β-arrestin1
monoclonal antibody was added at a 1:75 dilution (antibody: blocking buffer) and incubated at
room temperature for 2 hours. The cells were washed three times with PBS. The secondary
antibody (Jackson Immunoresearch Inc) was added (1:5000 dilution in blocking buffer) for 45
minutes at room temperature. Again the cells were washed three times with PBS and then
mounted using gel mount (Biomedia Corp, Foster City, CA) and microscope cover slips (Fisher
Scientific, Pittsburgh, PA). Z series images were obtained using a Bio-Rad MRC 1024 confocal
microscope. The acquired images were assembled using Adobe Photoshop and MS PowerPoint.
Co-Immunoprecipitations- Growth-arrested or transfected IIC9 cells were incubated in the
presence or absence of 1 unit/ml α-thrombin. Cell lysates were prepared and protein
concentrations were determined as mentioned above. Protein lysates (100 µg) were incubated
with 5 µg β-arrestin1 antibody or PI 3-Kinase p85 antibody at 4 °C with gentle rocking for 2
hours. The immune complexes were then immunoprecipitated as described previously (26),
resolved by SDS-polyacrylamide gel electrophoresis and transferred to a PVDF. Membranes
9
by guest on Decem
ber 25, 2019http://w
ww
.jbc.org/D
ownloaded from

were then probed with antibodies to β-arrestin1, PI 3-Kinase or G proteins (Calbiochem).
Immunoreactive bands were visualized by ECL detection.
PIP2 hydrolysis assay- PIP2 hydrolysis was determined as previously described (13).
10
by guest on Decem
ber 25, 2019http://w
ww
.jbc.org/D
ownloaded from

RESULTS
Rapid PI 3-Kinase activity is pertussis toxin-sensitive and mediated by Gβγ dimers - Previous
data from our laboratory (23) show that in IIC9 cells, α-thrombin activates rapid β-arrestin1-,
and Ras-dependent PI 3-Kinase activity. Because changes in Akt phosphorylation and activity
are dependent on PI 3-kinase activity (23), we quantified differences in Akt phosphorylation as a
measure of changes in PI 3-kinase activity. Similar to our reported results (23,24), α-thrombin
induces a 10-fold increase in Akt activity as determined by Western blot analysis using
antibodies directed against phosphorylated Akt. To determine whether the increase in PI 3-
kinase is dependent on a member of pertussis toxin (PTX) sensitive G proteins, we treated IIC9
cells with 100ng/ml of pertussis toxin for 6 hours prior to stimulation. Pretreatment with
pertussis toxin completely blocks the α-thrombin-induced increase in Akt phosphorylation (Fig.
1A) and PI 3-Kinase activity (Fig. 1C), indicating that α-thrombin-induced PI 3-kinase/Akt
pathway is dependent on a member of the Gi/o subfamily of G proteins. While pertussis toxin
blocks α-thrombin-stimulated Akt phosphorylation, it is ineffectual on Akt phosphorylation by
PDGF (Fig. 1A), which acts through a receptor tyrosine kinase and is insensitive to PTX. To
determine whether this activation is mediated by the Gα subunit or the Gβγ dimers, we
examined the effect of Gβγ sequestrants on the α-thrombin-induced activation of PI 3-Kinase
and Akt. Transient expression of both Gαt (Fig. 1B) and a membrane anchored GRK3 carboxyl-
terminal polypeptide, MAS-GRK3ct (data not shown) block α-thrombin-stimulated Akt
phosphorylation by over 65% whereas; expression of the vector alone is ineffectual (Fig. 1B).
Similar results are seen for PI 3-Kinase activity (Fig. 1C). Consistent with the known
mechanism of signaling through tyrosine kinase receptors, Gαt does not affect EGF-induced PI
3-Kinase activity (Fig. 1C). To ensure that the Gβγ sequestrants do not affect a Gα subunit
11
by guest on Decem
ber 25, 2019http://w
ww
.jbc.org/D
ownloaded from

mediated effect, we examined the result of expression of Gαt on PIP2 hydrolysis, which is
known to be mediated by Gαq (22). As previously found (13), addition of α-thrombin induces a
3- to 4-fold increase in inositol phosphate release (Fig. 3B). While expression of Gαt blocks Akt
phosphorylation (Fig. 1B), no change in the amount of thrombin-induced inositol phosphate
release is observed (Fig. 3B). Taken together, these data indicate that α-thrombin stimulates
rapid PI 3-kinase activity and Akt phosphorylation by the Gβγ dimers released from a PTX-
sensitive G protein.
α-Thrombin stimulates multiple heterotrimeric G proteins Gq, Gi2 and G13 - Several G protein
coupled receptors stimulate cellular responses by activating multiple G proteins that couple to
different signaling pathways. Because in many cell types, α-thrombin activates several G
proteins, including Gq/11, G13/12 and members of the pertussis toxin sensitive Gi subfamily,
we next investigated the G proteins activated by α-thrombin in IIC9 cells (Fig. 2). Previously we
found that IIC9 cells express Gαs, Gαi2, Gαi3, Gαo, Gαq/11 and Gα13 (21). We next examined
α-thrombin-induced binding of [35S]GTPγS to specific Gα subunits after immunoprecipitation
of the G proteins with Gα specific antibodies against Gαs, Gαi2, Gαi3, Gαo, Gαq, and Gα13
(27,28). Although significant binding of [35S]GTPγS occurs in unstimulated cells, α-thrombin
induces a 4-fold increase in [35S]GTPγS binding to Gαq, a 3-fold increase to Gα13 and an
approximate 5-fold increase to Gαi2 (Fig. 2A). α-Thrombin does not mediate significant binding
to Gαs, Gαi3 or Gαo (Fig. 2A). Because lysophosphatidic acid-mediated increases in Akt
phosphorylation are blocked by expression of Gα subunits C-terminal peptides to Gαo2 and not
Gαi2 (unpublished data), we reasoned that lysophosphatidic acid should couple to Go and not
Gi2. Consistent with the Akt phosphorylation data, lysophosphatidic acid stimulates a significant
increase in [35S]GTPγS binding to Gαo and not Gαi2, (Fig. 2B). Finally, treatment of isolated
12
by guest on Decem
ber 25, 2019http://w
ww
.jbc.org/D
ownloaded from

IIC9 membranes with the active peptide of cholera toxin induces detectable binding [35S]GTPγS
to only Gs (Fig. 2C). The thrombin-induced increases in [35S]GTPγS binding to Gαi2, Gαq, and
Gα13 indicate that α-thrombin couples to, and therefore can signal through, these G proteins.
Because some of the G proteins may be expressed at levels that are difficult to detect, and we
have not identified ligands that stimulate all of the G-proteins expressed, it is not possible to
determine the G-proteins activated by α-thrombin with certainty. However, these studies suggest
that α-thrombin couples primarily to Gi2, Gq and G13. Furthermore, we find no detectable
increases in [35S]GTPγS binding to other members of the pertussis toxin class as observed in
other cell types (19).
Gαq and Gαi2 are critical for α-thrombin-stimulated Akt phosphorylation - Because pertussis
toxin blocks α-thrombin-induced increase in Akt phosphorylation (Fig. 1A), and α-thrombin
induces a significant increase in GTPγS binding to Gi2 (Fig. 2), we reasoned that Gi2 mediates
PI 3-Kinase stimulation. To further demonstrate the involvement of Gi2, we expressed peptides
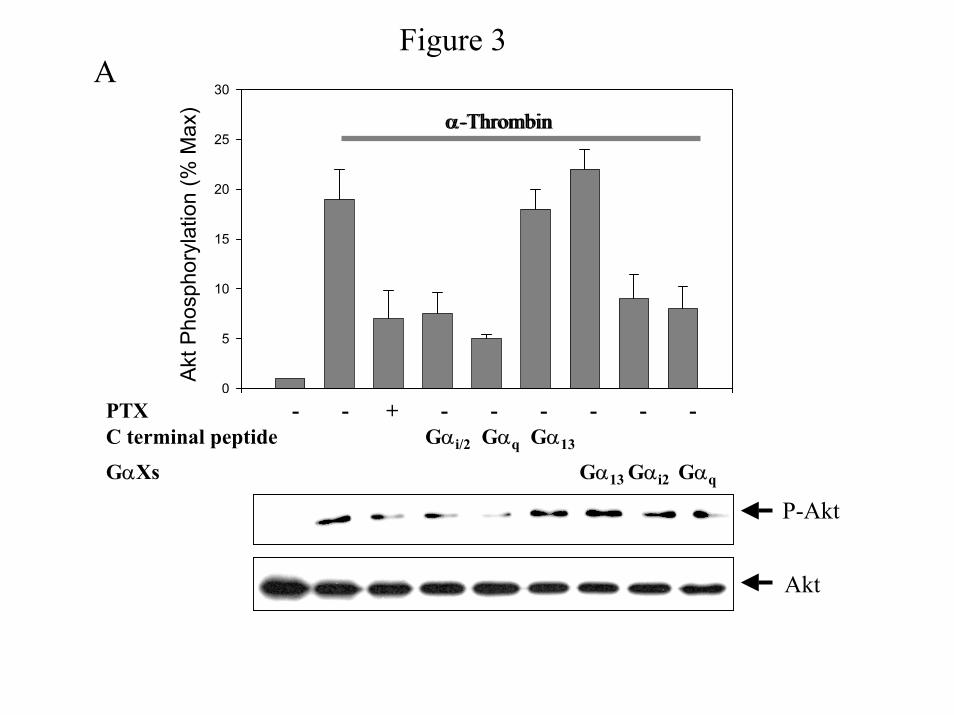
containing the C-terminus of Gα subunits (Fig. 3A). Expression of the carboxyl terminus Gα
peptides of specific Gα subunits inhibits binding and activation of the targeted Gα subunit
(14,29), indicating that these peptides effectively inhibit specific GPCR-mediated responses.
Consistent with the data in Figures 1 and 2, expression of the C-terminal peptide of Gαi1/2
inhibits α-thrombin-induced Akt phosphorylation (Fig. 3A). Furthermore, expression of Gα13
(Fig. 3A), Gα01, Gα02 (data not shown) is ineffective. Surprisingly, rapid Akt phosphorylation
is inhibited by expression of the C-terminal peptide of Gαq (Fig. 3A), suggesting that α-
thrombin-induced PI 3-Kinase activation is dependent on Gαq. We obtained similar results for
PI 3-kinase activity (data not shown). The unexpected activity of the C-terminal peptide of Gαq
prompted us to characterize these peptides further. We reasoned that PIP2 hydrolysis and Rho
13
by guest on Decem
ber 25, 2019http://w
ww
.jbc.org/D
ownloaded from

activity would be dependent on Gq and G13 (14), respectively. While expression of the Gαq C-
terminal peptide blocks PIP2 hydrolysis, expression of Gαi1/2 C-terminal peptide or PTX
treatment does not (Fig. 3B). Furthermore, only expression of the C-terminal peptide of Gα13
inhibits Rho activation, while Gαi1/2 and Gαq are ineffective (Fig. 3C). We also measured the
ability of the C-terminal peptides to inhibit sustained ERK phosphorylation, which is
dependent on PTX-sensitive G proteins (Gi2). We find that expression of the C-terminal
peptide of Gαi1/2 inhibits sustained ERK activation, while Gα13 and Gαq do not inhibit it (Fig.
3C). Taken, together these data indicate that the C-terminal peptides effectively inhibit targeted
Gα subunits and that Akt phosphorylation is dependent on both Gq and Gi2.
We were surprised that Akt phosphorylation is sensitive to uncoupling of Gαq
from α-thrombin. To ensure that these results are not selective for C-terminal peptides, we
decided to use another approach to inhibit G protein activation, utilizing xanthine nucleotide
binding Gα mutants (GαXs). Expression of GαX inhibits coupling of GPCR, including the
thrombin receptor, to specific G proteins indicating that these GαXs act as dominant negative
inhibitors (30). We transiently transfected IIC9 cells with Gαi2X, GαqX and Gα13X, and
quantified Akt phosphorylation (Fig. 3A). Similar to the results with the C-terminal peptides,
Gαi2X and GαqX block Akt phosphorylation, while Gα13X does not. Consistent with the ability
of these GαX constructs to inhibit the activation of specific G proteins by α-thrombin,
expression of GαqX blocks and Gαi2X does not inhibit PIP2 hydrolysis (data not shown).
Taken together, these data show that α-thrombin-induced PI 3-Kinase activity and Akt
phosphorylation is dependent on both Gq and Gi2.
Expression of Pertussis toxin-insensitive Gi2α rescues α-thrombin-stimulated Akt
phosphorylation in the presence of pertussis toxin – Because our data show that α-thrombin-
14
by guest on Decem
ber 25, 2019http://w
ww
.jbc.org/D
ownloaded from

induced Akt phosphorylation is dependent on both Gq and Gi2, we reasoned that expression of
a pertussis toxin-resistant mutant of Gαi2 should rescue pertussis toxin inhibition of α-
thrombin-induced Akt phosphorylation. Pertussis toxin-resistant mutants were generated by
converting the C-terminal of Gα from cysteine residue to a glycine (GαoC351G, Gαi2C352G,
and Gαi3C351G). To verify that the C→G mutation renders the expressed Gα subunits
resistant to pertussis toxin treatment, IIC9 cells were transiently transfected with vector alone
or vector containing GαoC351G, Gαi2C352G, or Gαi3C351G, treated with 100ng/ml pertussis
toxin for 10 hours and then cell lysates were analyzed by SDS-PAGE in the presence of 6M
urea (Fig. 4A). Previous data in IIC9 cells show that pre-treatment with 100ng/ml pertussis
toxin for 10 hours results in the ADP-ribosylation of more than 95% of the pertussis toxin-
sensitive G proteins (22). For each construct, in the absence of pertussis toxin western blot
analysis with an antibody that recognizes all Gi family members shows a single band with
apparent molecular weight of 43kDa (Fig. 4A). Treatment with pertussis toxin results in a
single band shifted to higher molecular weight (Fig 4B). In the presence of pertussis toxin,
western blot analysis of lysate proteins from cells expressing each construct shows a band at
43kDa and a faster migrating band (Fig 4B). Presumably, the lower band is the unmodified Gα
subunit and is absent in lysates from cells transfected with empty vector. Furthermore, transient
expression of the three Gα C→G constructs results in similar protein levels (Fig. 4B) ensuring
that the ability to rescue is independent of expression levels. These data clearly demonstrate
that treatment with pertussis toxin for 10 hours results in ADP-ribosylation of the endogenous
Gi family members and the Gα C→G mutants are resistant to pertussis toxin. In agreement
with our data indicating a role for Gi2 in α-thrombin-stimulated Akt phosphorylation,
expression of Gαi2C352G but not GαoC351G or Gαi3C351G rescues α-thrombin-induced rapid
Akt phosphorylation in IIC9 cells treated with pertussis toxin (Fig. 4C).
15
by guest on Decem
ber 25, 2019http://w
ww
.jbc.org/D
ownloaded from

α-Thrombin induced translocation of β-arrestin1 to the plasma membrane is mediated by Gβγ
released from Gαq - Because β-arrestin1 is required for thrombin-induced PI 3-Kinase and Akt
activities in IIC9 cells (23), we addressed the possibility that β-arrestin1 translocation is
dependent on a single G-protein. We have previously established that treatment of IIC9 cells
with α-thrombin results in the translocation of β-arrestin1 to the plasma membrane (23). We
have also shown that expression of a membrane anchored GRK3 carboxyl-terminal polypeptide
(MAS-GRK3ct) blocks the translocation, indicating the involvement of a Gβγ (23). We next
examined the effect of expression of C-terminal peptides on α-thrombin-induced translocation
of β-arrestin1 (Fig. 5) by both biochemical fractionation (Fig. 5A) and confocal microscopy
(Fig. 5B&C). We found it necessary to transiently express β-arrestin1 to examine the
translocation. α-Thrombin induces a significant increase in the translocation of β-arrestin1 to
the membrane fraction within 5 minutes (Fig. 5A) and in the translocation of a substantial
fraction of the cellular fluorescence to the plasma membrane within 10-30 minutes (Fig. 5B,
2&3). After 30 minutes, β-arrestin1 is found mainly in the cytoplasm (Fig. 5B, 4). While
expression of the C-terminal peptide of Gαq inhibits translocation (Fig. 5A & 5C, 4), treatment
with PTX or expression of the C-terminal peptide of Gαi1/2 does not block the amount of β-
arrestin1 translocation (Fig. 5A & 5C, 3&6). However, transient co-expression of Gαt blocks
the increase (Fig. 5A) and also results in an evenly distributed cytoplasmic fluorescence (Fig.
5C, 5). These data indicate that β-arrestin1 translocation to the plasma membrane in IIC9 cells
is mediated by the Gβγ released from Gαq and suggest that Gi2 affects PI 3-Kinase activation
by another mechanism.
α-Thrombin-mediated formation of complexes containing PI 3-Kinase and β-arrestin1 -
Because the ability of β-arrestin1 to associate with members of MAPK pathways is known to
16
by guest on Decem
ber 25, 2019http://w
ww
.jbc.org/D
ownloaded from

affect activation of these pathways (31), we reasoned that β-arrestin1 serves as an adapter
molecule, associating with and recruiting PI 3-Kinase to the membrane. To determine whether
α-thrombin induces association of β-arrestin1 with PI 3-Kinase, we immunoprecipitated β-
arrestin1 pre and post α-thrombin stimulation and examined the immunoprecipites for the
presence of PI 3-Kinase (Fig. 6). In the absence of α-thrombin, low amounts of PI 3-Kinase are
found in the immunoprecipitates (Fig. 6). α-Thrombin induces an increase of PI 3-Kinase in β-
arrestin1 immunoprecipitates within 15 minutes that decreases to amounts similar to those
found in unstimulated cells after 30 minutes (data not shown). Similar results are found when
PI 3-Kinase immunoprecipitates are assayed for the presence of β-arrestin1 (data not shown).
Expression of Gαq C-terminal peptide inhibits complex formation while the Gαi1/2 C-terminal
peptide does not (Fig. 6). Taken together, these data indicate that α-thrombin stimulates
membrane translocation of β-arrestin1 and its association with PI 3-Kinase suggesting that, as
is found in MAPK pathway activation (32), β-arrestin1 functions as an adaptor molecule
recruiting PI 3-Kinase.
Gi2 mediates PI 3-Kinase activation by α-thrombin via Ras - While our results clearly
demonstrate a role for Gq in the activation of PI 3-kinase, the data do not provide a mechanistic
role for Gi2. Previously we found that Ras is required for α-thrombin-induced PI 3-Kinase
activation (24). Furthermore, Ras stimulation by α-thrombin is independent of β-arrestin1 (23),
suggesting that Ras activation is independent of Gq. These data suggest that Gi2 may affect PI
3-Kinase via Ras. To test this hypothesis, we next examined the role of G proteins in the
activation of α-thrombin-induced Ras activation using the RBD fragment of the Ras effector
Raf-1 (33). Treatment of IIC9 cells with α-thrombin results in an approximate 12-fold increase
of GTP-bound Ras (Fig. 7). The α-thrombin-induced increase is blocked by pretreatment of IIC9
17
by guest on Decem
ber 25, 2019http://w
ww
.jbc.org/D
ownloaded from

cells with pertussis toxin or expression of the Gαi1/2 C-terminal peptide (Fig. 7A). In contrast to
the ability of the Gαi1/2 C-terminal peptide to block the Ras activation, expression of the Gαq C-
terminal peptide does not affect the increase (Fig. 7A). Also we find that expression of pertussis
toxin-insensitive Gαi2C352G but not GαoC351G nor Gαi3C351G rescues α-thrombin-induced
Ras activation in IIC9 cells treated with PTX (Fig. 7B), confirming the ability of the C-terminal
peptide of Gαi1/2 to block Ras activation.
Because GPCRs that are sensitive to pertussis toxin often stimulate Ras through
Gβγ subunits (5), we reasoned that expression of Gβγ sequestrants would block Ras activation
by α-thrombin. Expression of Gαt completely abolishes α-thrombin mediated GTP-binding to
Ras (Fig. 7A). Taken together, these data demonstrate the involvement of Gβγ dimers from Gαi2
in α-thrombin-stimulated Ras and thus a role in PI 3-Kinase activation.
18
by guest on Decem
ber 25, 2019http://w
ww
.jbc.org/D
ownloaded from

DISCUSSION
α-Thrombin stimulates multiple effectors in fibroblasts (1,22,34), including PI-
PLC, ERK and PI 3-Kinase activities. In this paper, we identify the G proteins that mediate the
activation of PI 3-Kinase and Akt by α-thrombin. We find that treatment of IIC9 cells with PTX
markedly inhibits PI 3-Kinase and Akt activation in response to α-thrombin (Fig. 1). Because
PTX catalyzes the ADP-ribosylation and inactivation of members of the Gi/o family (9), these
data indicate the involvement of Gi/o. Consistent with these data, α-thrombin activates the PTX-
sensitive G-protein, Gi2 (Fig. 2). We then reasoned that Gi2 mediates PI 3-Kinase and Akt
activation in response to α-thrombin. Consistent with this notion, expression of either Gαi1/2 C-
terminal blocking peptide (29) or dominant negative Gαi2X mutant (30) blocks Akt
phosphorylation and PI 3-Kinase activity (Fig. 3). Surprisingly, expression of a Gαq C-terminal
peptide, which blocks the activation of Gq, or the dominant negative GαqX mutant, inhibits Akt
phosphorylation (Fig. 3A), indicating an important role for Gq in PI 3-Kinase activation by α-
thrombin.
Previously, we reported that β-arrestin1 is required for α-thrombin-induced PI 3-
Kinase and Akt activities in IIC9 cells (23,35). However, we had not identified the G proteins
that mediate activation of the PI 3-Kinase/Akt pathway. Expression of a dominant negative
GαqX mutant and a Gαq C-terminal peptide blocks β-arrestin1 translocation, while expression of
dominant negative Gαi2X mutant or the Gαi1/2 C-terminal peptide is ineffective (Fig. 5).
Furthermore, β-arrestin1 translocation is dependent on the Gβγ subunits of Gαq, indicating an
essential role for Gβγ in β-arrestin1 translocation.
In addition to Gq, there is also a role for Gi2 in α-thrombin-induced PI 3-Kinase
and Akt stimulation. Our results demonstrate that Ras is upstream of PI 3-Kinase activation and
19
by guest on Decem
ber 25, 2019http://w
ww
.jbc.org/D
ownloaded from

the dependence of PI 3-Kinase on Ras is unrelated to the dependence of PI 3-Kinase on β-
arrestin1. First, inhibition of Gαi2 activation abrogates Ras activation (Fig. 7A). Second,
expression of only PTX-resistant Gαi2 rescues α-thrombin-induced Ras activation in PTX-
treated IIC9 cells (Fig. 7B). Furthermore, α-thrombin induces Ras via the Gβγ dimers released
from Gαi2 (Fig. 7A). Taken together, these results indicate that α-thrombin stimulates Ras via
the Gβγ from Gi2.
We were surprised to find that the translocation of β-arrestin1 and the activation of
Ras were mediated by the Gβγ subunits from Gq and Gi2, respectively. These results show that
the Gβγs associated with Gq and Gi2 can activate different effectors. There are several possible
explanations to account for the specificity of Gβγ signaling of Gq and Gi2. It is possible that Gαq
and Gαi2 associate with different subsets of Gβγs, and the distinct subsets activate distinct
effectors. Several studies lend support to the notion that select Gβγ subunits activate distinct
effectors (36). Co-transfection experiments in COS-7 cells found that expression of only specific
Gβ or Gγ subunits significantly activate PI-PLCβ2 (37). Whereas expression of Gβ1γ1, Gβ1γ5 or
Gβ2γ5 stimulates PI-PLCβ2 activity, expression of Gβ2γ1 has no effect. Similarly, expression of
Gβ5γ2 stimulates PI-PLBβ2 but not ERK, while expression of Gβ1γ2 activates both (38). Other
studies (39) found that expression of only specific Gβ subunits activate G protein receptor
kinases2 (GRK2). Of the Gβ subunits examined, Gβ1 and Gβ2 but not Gβ3 activates GRK2.
Recent data using immobilized Gβ1γ2 to screen phage-displayed random peptide libraries (40)
found a peptide, which contains a conserved sequence found in PI-PLCβ. Interestingly,
expression of this peptide blocks activation of PI-PLCβ, but not Gβγ dependent voltage-gated
channels or Gβγ-mediated inhibition of type I adenylate cyclase suggesting that effectors bind to
specific Gβγ peptide sequences (40).
20
by guest on Decem
ber 25, 2019http://w
ww
.jbc.org/D
ownloaded from

For different G-proteins to activate distinct effectors via release of Gβγ suggests
that the Gαs are associated with distinct subsets of Gβγs. While there is strong evidence for the
activation of effectors by specific Gβγ subunits, there is little data on the identity of the Gβγ
associated with specific Gαs. Consistent with this requirement, analysis of the Gβ subunits
associated with Gαi2 and Gαq in IIC9 cells (Reema, G and Baldassare, JJ, unpublished results)
find different subsets of Gβs associated with Gαi2 and Gαq.
A second explanation for our results is that activation of Ras and translocation of
β-arrestin1 occurs in different membrane domains, for example in raft and non-raft membrane
fractions (41-44). These membrane domains are enriched in proteins important in intracellular
signaling including GPCRs, suggesting that these domains may play a role in signaling from
GPCRs to their effectors. Huang et al (45) have reported enrichment of Gi, Gs, Go and Gβγs
in detergent-resistant membrane domains, lipid rafts . Furthermore, H-Ras has been shown to
be localized to rafts, suggesting that Ras activation could occur in these domains (46).
At present our data cannot distinguish between these mechanisms. However, any
mechanism must take into account our results showing that the activation of PI 3-kinase by α-
thrombin (Fig. 8) is mediated via Gq and Gi2. Gq plays a crucial role in mediating α-thrombin-
induced PI 3-kinase activation through β-arrestin1 by stimulating β-arrestin1 association with
PI 3-kinase, suggesting that β-arrestin1 functions as an adaptor molecule recruiting PI 3-kinase.
PI 3-kinase activation also requires Ras activation, which is downstream of Gi2. Both effectors
are blocked by Gβγ sequestrants, indicating that they are Gβγ-regulated. Because Ras
activation and β-arrestin1 translocation are crucial for α-thrombin-mediated PI 3-kinase
activity and, therefore, likely occur in the same domain, we think that the association of
different Gβγs with specific Gαs is likely essential. However, it is also important to keep in
21
by guest on Decem
ber 25, 2019http://w
ww
.jbc.org/D
ownloaded from

mind that these mechanisms are not mutually exclusive. Thus, both could play a role. The
possible importance of these mechanisms in Gβγ signaling remains to be elucidated.
22
by guest on Decem
ber 25, 2019http://w
ww
.jbc.org/D
ownloaded from

References
1. Chambard, J., Paris, S., A'llemain, G., and Pouysségur, J. (1987) Nature 326, 800-803
2. Coughlin, S. R. (1999) Proc.Natl.Acad.Sci.U.S.A. 96, 11023-11027
3. Bourne, H. R. (1997) Curr.Opin.Cell Biol. 9, 134-142
4. Hawes, B. E., van Biesen, T., Koch, W. J., Luttrell, L. M., and Lefkowitz, R. J. (1995) Journal of Biological Chemistry 270, 17148-17153
5. Crespo, P., Xu, N. Z., Simonds, W. F., and Gutkind, J. S. (1995) Journal of Cellular Biochemistry 13
6. Brock, C., Schaefer, M., Reusch, H. P., Czupalla, C., Michalke, M., Spicher, K., Schultz, G., and Nurnberg, B. (2003) Journal of Cell Biology 160, 89-99
7. Kurosu, H., Maehama, T., Okada, T., Yamamoto, T., Hoshino, S., Fukui, Y., Ui, M., Hazeki, O., and Katada, T. (1997) Journal of Biological Chemistry 272, 24252-24256
8. Simon, M. I., Strathmann, M. P., and Gautam, N. (1991) Science 252, 802-808
9. Fields, T. A. and Casey, P. J. (1997) Biochemical Journal 321, 561-571
10. Dhanasekaran, N. and Prasad, M. V. (1998) Biol.Signals Recept. 7, 109-117
11. Aragay, A. M., Collins, L. R., Post, G. R., Watson, A. J., Feramisco, J. R., Brown, J. H., and Simon, M. I. (1995) Journal of Biological Chemistry 270, 20073-20077
12. Baffy, G., Yang, L., Raj, S., Manning, D. R., and Williamson, J. R. (1994) J.Biol.Chem. 269, 8483-8487
13. Gardner, A., Phillips-Mason, P. J., Raben, D. M., and Baldassare, J. J. (2002) Cellular Signalling 14, 499-507
14. Gilchrist, A., Vanhauwe, J. F., Li, A. L., Thomas, T. O., Voyno-Yasenetskaya, T., and Hamm, H. E. (2001) Journal of Biological Chemistry 276, 25672-25679
15. Kanthou, C., Kanse, S. M., Kakkar, V. V., and Benzakour, O. (1996) Cellular Signalling 8, 59-66
23
by guest on Decem
ber 25, 2019http://w
ww
.jbc.org/D
ownloaded from

16. Ogino, Y., Tanaka, K., and Shimizu, N. (1996) European Journal of Pharmacology 316, 105-109
17. Post, G. R., Collins, L. R., Kennedy, E. D., Moskowitz, S. A., Aragay, A. M., Goldstein, D., and Brown, J. H. (1996) Mol.Biol.Cell 7, 1679-1690
18. Rahman, A., True, A. L., Anwar, K. N., Ye, R. D., Voyno-Yasenetskaya, T. A., and Malik, A. B. (2002) Circulation Research 91, 398-405
19. Vanhauwe, J. F., Thomas, T. O., Minshall, R. D., Tiruppathi, C., Li, A. L., Gilchrist, A., Yoon, E., Malik, A. B., and Hamm, H. E. (2002) Journal of Biological Chemistry 277, 34143-34149
20. Verrall, S., Ishii, M., Chen, M., Wang, L., Tram, T., and Coughlin, S. R. (1997) Journal of Biological Chemistry 272, 6898-6902
21. Cheng, J., Weber, J. D., Baldassare, J. J., and Raben, D. M. (1997) Journal of Biological Chemistry 272, 17312-17319
22. Gardner, A. J. (1998) Dissertation 1-2
23. Goel, R., Phillips-Mason, P. J., Raben, D. M., and Baldassare, J. J. (2002) Journal of Biological Chemistry 277, 18640-18648
24. Phillips-Mason, P. J., Raben, D. M., and Baldassare, J. J. (2000) Journal of Biological Chemistry 275, 18046-18053
25. Weber, J. D., Cheng, J., Raben, D. M., Gardner, A., and Baldassare, J. J. (1997) Journal of Biological Chemistry 272, 17320-17326
26. Lents, N. H., Keenan, S. M., Bellone, C., and Baldassare, J. J. (2002) Journal of Biological Chemistry 277, 47469-47475
27. Barr, A. J., Brass, L. F., and Manning, D. R. (1997) Journal of Biological Chemistry 272, 2223-2229
28. Windh, R. T. and Manning, D. R. (2002) G Protein Pathways Part B: G Proteins and Their Regulators 344, 3-14
29. Gilchrist, A., Bunemann, M., Li, A., Hosey, M. M., and Hamm, H. E. (1999) Journal of Biological Chemistry 274, 6610-6616
30. Yu, B., Gu, L. J., and Simon, M. I. (2000) Journal of Biological Chemistry 275, 71-76
31. Pierce, K. L., Luttrell, L. M., and Lefkowitz, R. J. (2001) Oncogene 20, 1532-1539
32. Miller, W. E. and Lefkowitz, R. J. (2001) Curr.Opin.Cell Biol. 13, 139-145
24
by guest on Decem
ber 25, 2019http://w
ww
.jbc.org/D
ownloaded from

33. Taylor, S. J., Resnick, R. J., and Shalloway, D. (2001) Regulators and Effectors of Small Gtpases, Pt G 333, 333-342
34. Grand, R. J. A., Turnell, A. S., and Grabham, P. W. (1996) Biochemical Journal 313, 353-368
35. Goel, R. and Baldassare, J. J. (2002) Cell Signaling, Transcription, and Translation As Therapeutic Targets 973, 138-141
36. Albert, P. R. and Robillard, L. (2002) Cellular Signalling 14, 407-418
37. Wu, D., Katz, A., and Simon, M. I. (1993) Proc.Natl.Acad.Sci.U.S.A. 90, 5297-5301
38. Zhang, S., Coso, O. A., Lee, C., Gutkind, J. S., and Simonds, W. F. (1996) J.Biol.Chem. 271, 33575-33579
39. Daaka, Y., Pitcher, J. A., Richardson, M., Stoffel, R. H., Robishaw, J. D., and Lefkowitz, R. J. (1997) Proc.Natl.Acad.Sci.U.S.A. 94, 2180-2185
40. Scott, J. K., Huang, S. F., Gangadhar, B. P., Samoriski, G. M., Clapp, P., Gross, R. A., Taussig, R., and Smrcka, A. V. (2001) Embo Journal 20, 767-776
41. Brown, D. A. and London, E. (1998) Annu.Rev.Cell Dev.Biol. 14, 111-136
42. Dobrowsky, R. T. (2000) Cell Signal. 12, 81-90
43. Heximer, S. P., Srinivasa, S. P., Bernstein, L. S., Bernard, J. L., Linder, M. E., Hepler, J. R., and Blumer, K. J. (1999) J.Biol.Chem. 274, 34253-34259
44. Kurzchalia, T. V. and Parton, R. G. (1999) Curr.Opin.Cell Biol. 11, 424-431
45. Huang, C., Hepler, J. R., Chen, L. T., Gilman, A. G., Anderson, R. G., and Mumby, S. M. (1997) Mol.Biol.Cell 8, 2365-2378
46. Prior, I. A., Harding, A., Yan, J., Sluimer, J., Parton, R. G., and Hancock, J. F. (2001) Nat.Cell Biol. 3, 368-375
25
by guest on Decem
ber 25, 2019http://w
ww
.jbc.org/D
ownloaded from

Footnotes
1The abbreviations used are: IIC9 cells, Chinese hamster embryonic fibroblasts; GPCR,
G-protein coupled receptor; Proteinase Activated Receptor, PAR; α-transducin, Gαt; PI3-
Kinase, phosphatidylinositol-3OH-Kinase; ERK, extracellular-related kinase; PAGE,
polyacrylamide gel electrophoresis; PLC, phospholipase C; PKC, protein kinase C; GTPγS,
guanosine-5’-O-(3-thio)triphosphate; Polyvinylidene difluoride membrane, PVDF.
This work is supported by GM59251(DMR)
†Present address is Department of Molecular Biology and Microbiology, Case Western
Reserve University, Cleveland, Ohio 44106.
±Present address is Department of Pharmaceutical Sciences, Massachusetts College of
Pharmacy & Health Sciences, Worcester, MA, 01608.
26
by guest on Decem
ber 25, 2019http://w
ww
.jbc.org/D
ownloaded from

FIGURE LEGENDS
Figure 1: PI 3-Kinase pathway is pertussis toxin-sensitive and mediated by Gβγ dimers.
Cells were untreated or transfected with pcDNA (Invitrogen) or pcDNA containing Gαt. Growth
arrested IIC9 cells were untreated or incubated in the presence of 100ng/ml PTX 6 hours prior to
stimulation with the indicated mitogen for 5 minutes (1 unit/ml α-thrombin or 10 ng/ml PDGF
or 10 ng/ml EGF). A) & B) Lysate proteins (20 µg) were separated by SDS-PAGE,
immunoblotted with either anti-Akt or anti-phospho-Akt polyclonal antibody and the blots
quantified using a Molecular Dynamics densitometer. Relative activation of Akt (p-Akt /Akt) is
shown, with activity of serum arrested IIC9 as 1.0. Results for both A and B are representative
blots of four independent experiments. C) PI 3-Kinase complexes were immunoprecipitated
from lysates containing equal proteins using an anti-p85 polyclonal antibody and assayed for the
ability to phosphorylate PI in vitro. PI 3-Kinase activity was quantified as previously described
(1). The data shown are the mean ± S. D. for triplicates in one experiment and are representative
of four independent experiments. The Western blot is a representative blot.
Figure 2: α-Thrombin stimulates the binding of [35S]GTPγS to Gq, Gi2 and G13.
Membranes were prepared from serum arrested IIC9 cells and the G protein immune complexes
containing specific G proteins were immunoprecipitated as described in “Materials and
Methods” after treatment with A) 1 unit/ml thrombin or B) 20µM LPA or C) 1µg/assay of
cholera toxin. The presence of Gα subunits was analyzed by Western blot analysis using the
indicated Gα specific antibodies. In control samples, membranes proteins were
immunoprecipitated with anti-Gα subunit antibody (Calbiochem, CA). [35S]GTPγS binding in
the immunoprecipitates was quantified by scintillation counting. The data shown are the mean
± S. D. for triplicates in one experiment and are representative of four independent experiments.
27
by guest on Decem
ber 25, 2019http://w
ww
.jbc.org/D
ownloaded from

Figure 3: Effect of C-terminal peptides of Gα subunits on α-thrombin-stimulated
effectors. IIC9 cells were untreated or transfected with the C-terminal peptides of Gα subunits
and then serum arrested. A) Inhibition of Gαq or Gαi2 activation blocks α-thrombin-
stimulated Akt phosphorylation. Cells were transfected with the GαXs mutants or the C-
terminal peptides as indicated in figure. The serum arrested cells were untreated or treated with
100ng/ml PTX for 6 hours prior to incubation in the absence or presence of 1 unit/ml α-
thrombin for 5 minutes. Cell lysates (20 µg) were separated by SDS-PAGE, and immunoblotted
with either anti-Akt or anti-phospho-Akt polyclonal antibody. Blots were quantified using a
Molecular Dynamics densitometer. The data shown are the mean ± S. D. for triplicates in one
experiment and are representative of three independent experiments. The Western blot is a
representative blot. B) Effect of C-terminal peptides on PIP2 hydrolysis. Serum arrested
cells were labeled with myo-[3H]inositol (1 µCi/ml) for 24 hours, and then incubated in serum
free medium supplemented with 20 mM LiCl in the absence or presence of 1 Unit/ml α-
thrombin for 20 minutes at 37oC, and IP3 amounts quantified. The data shown are the mean ±
S. D. for duplicates in one experiment and are representative of three independent experiments.
C) Effect of C-terminal peptides on Rho activity and sustained ERK phosphorylation.
Serum arrested cells were incubated in the absence or presence of 1 unit/ml α-thrombin for 5
minutes and 4 hours for Rho activity and sustained ERK phosphorylation, respectively. For
Rho activity, cells lysates were prepared, incubated with recombinant GST-RBD for 1 hour,
and the immucomplexes subjected to SDS-PAGE and Western blot analysis with anti-RhoA
antibody. For ERK activity the cell lysates were subjected to SDS-PAGE and Western blot
analysis with anti-phospho-ERK polyclonal antibody. Relative activation of RhoA (complexed
with GTP) and ERK are shown in the same graph. We calculated these activities in the
28
by guest on Decem
ber 25, 2019http://w
ww
.jbc.org/D
ownloaded from

following manner. We subtracted the basal activities (for Rho and Phospho-ERK) from the α-
thrombin-induced activities in the untransfected cells and set these values equal to 100%. The
transfected samples are calculated by subtracting the basal from the experimental and dividing
these values by the untransfected values times 100. The data shown are representative of three
independent experiments.
Figure 4: C G Gi Mutants Are Not ADP-ribosylated: Cells were transiently transfected
with either vector (pcDNA), or pcDNA containing GoαC351G, Gi2αC352G, and Gi3αC351G.
The transfected cells were growth arrested, and cell lysates prepared from: A) cells treated with
100 ng/ml PTX for 6 hours prior to the preparation of lysates or B) untreated cells. Lysate
proteins (20 µg) were separated by SDS-PAGE containing 6M urea and immunoblotted with a
polyclonal antibody to an internal sequence recognized by Gα subunits. C) Expression of PTX-
insensitive Gαi2 rescues α-thrombin-stimulated Akt phosphorylation in IIC9 cells treated
with PTX. The transfected cells were incubated in the presence or absence of 100 ng/ml
pertussis toxin 6 hours prior to stimulation with 1 unit/ml α-thrombin for 5 minutes and cell
lysates prepared. Lysate proteins (20 µg) were immunoblotted with anti-phospho-Akt or anti-
Akt polyclonal antibody. Akt phosphorylation was quantified using a Molecular Dynamic
PhosphoimagerTM and reported as percent maximal stimulation. Data in A) and B) are
representative of three independent experiments. The data shown in C) are the mean ± S. D. for
triplicates in one experiment and are representative of four independent experiments. The
Western blot is a representative blot.
Figure 5: Expression of Gβγ sequestrant or C-terminal Gαq blocks α-thrombin induced
translocation of β-arrestin1. IIC9 cells were transfected with β-arrestin1 alone or with Gαt or
29
by guest on Decem
ber 25, 2019http://w
ww
.jbc.org/D
ownloaded from

the C-terminal peptides of Gαq or Gαi1/2. The cells were then stimulated with 1 unit/ml α-
thrombin. A) Membranes were prepared after stimulation for 15 minutes. Membrane protein
lysates (50 µg) were separated by SDS-PAGE, immunoblotted, and the amount of β-arrestin1
quantified with a Molecular Dynamics densitometer. The data shown are the mean ± S. D. for
triplicates in one experiment and are representative of three independent experiments. The
Western blot is a representative blot. B) & C) IIC9 cells were grown on chamber slides and
transfected as described above. The cells were fixed, permeabilized and visualized by confocal
microscopy as described under “Material and Methods”. B) β-Arrestin1 was visualized after 0
minutes, 10 minutes, 30 minutes and 1 hour post stimulation with α-thrombin. C) The cells were
serum arrested, and were untreated or treated for 4 hour with 100 ng of PTX. After treatment the
cells were stimulated with 1 unit/ml α-thrombin for 30 min. Data in B and C are representative
of three independent experiments.
Figure 6: α-Thrombin-mediated formation of complexes containing PI 3-Kinase and β-
arrestin1. IIC9 cells were transiently co-transfected with 1µg/ml β-arrestin1 and C-terminal
peptides of Gαi1/2 or Gαq. The cells were serum arrested, and treated with or without 1 unit/ml of
α-thrombin for 15 minutes. Protein lysates (100 µg) were incubated with antibodies directed
against β-arrestin1 or PI 3-Kinase p85 for 2 hours. The immune complexes were
immunoprecipitated as described in “Materials and Methods”. Samples were subjected to SDS-
PAGE and probed with the indicated antibodies directed against β-arrestin1or PI 3-Kinase p85
and the amount of β-arrestin1 or PI 3-Kinase p85 quantified with a Molecular Dynamics
densitometer. This is a representative blot of five independent experiments
30
by guest on Decem
ber 25, 2019http://w
ww
.jbc.org/D
ownloaded from

31
Figure 7: Gi2 mediates Ras activation. Cells were transfected with A) Gαt or the C-terminal
peptides Gαi1/2 or Gαq or B) pcDNA containing GαoC351G, Gαi2C352G, and Gαi3C351G. The
cells were growth arrested. Growth arrested cells were incubated in the presence or absence of
100ng/ml PTX 6 hours prior to stimulation and then incubated for 15 minutes in the absence or
presence of 1 unit/ml α-thrombin. Cells lysates were prepared and incubated with recombinant
GST-RBD for 1 hour. Samples were subjected to SDS-PAGE, probed with anti-pan-Ras
antibody and the amounts of Ras quantified. The data shown in A) are the mean ± S. D. for
triplicates in one experiment and are representative of three independent experiments. The
Western blot is a representative blot. Data in B) is representative of two independent
experiments.
Figure 8: Signal transduction pathway involved in α-thrombin-induced PI 3-Kinase.
by guest on Decem
ber 25, 2019http://w
ww
.jbc.org/D
ownloaded from
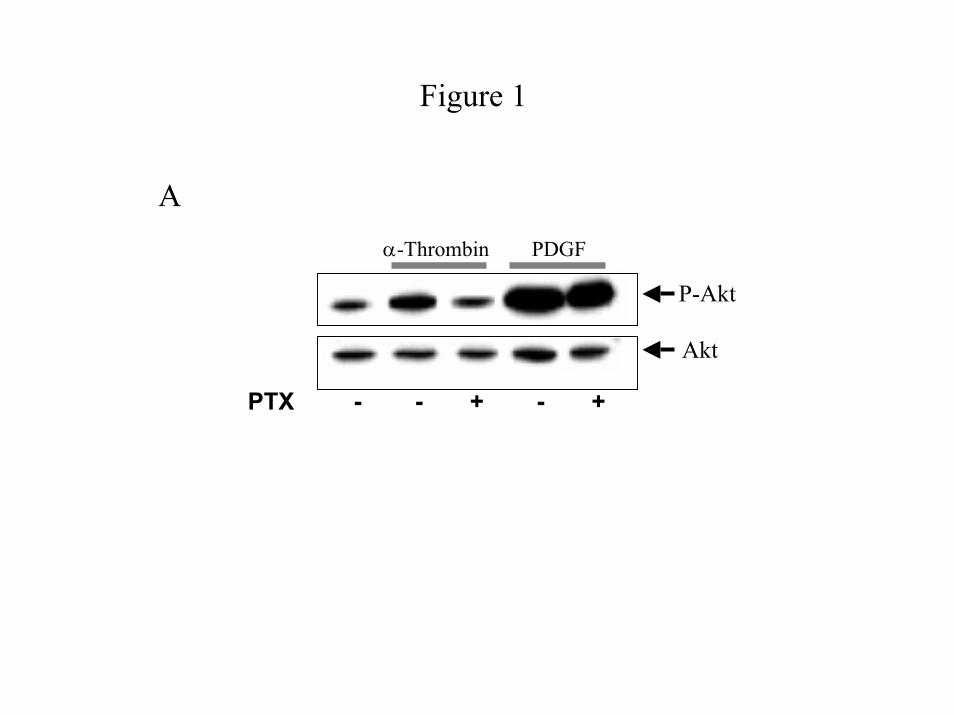
Figure 1
A
P-Akt
Akt
α-Thrombin PDGF
PTX - - + - +
by guest on December 25, 2019 http://www.jbc.org/ Downloaded from
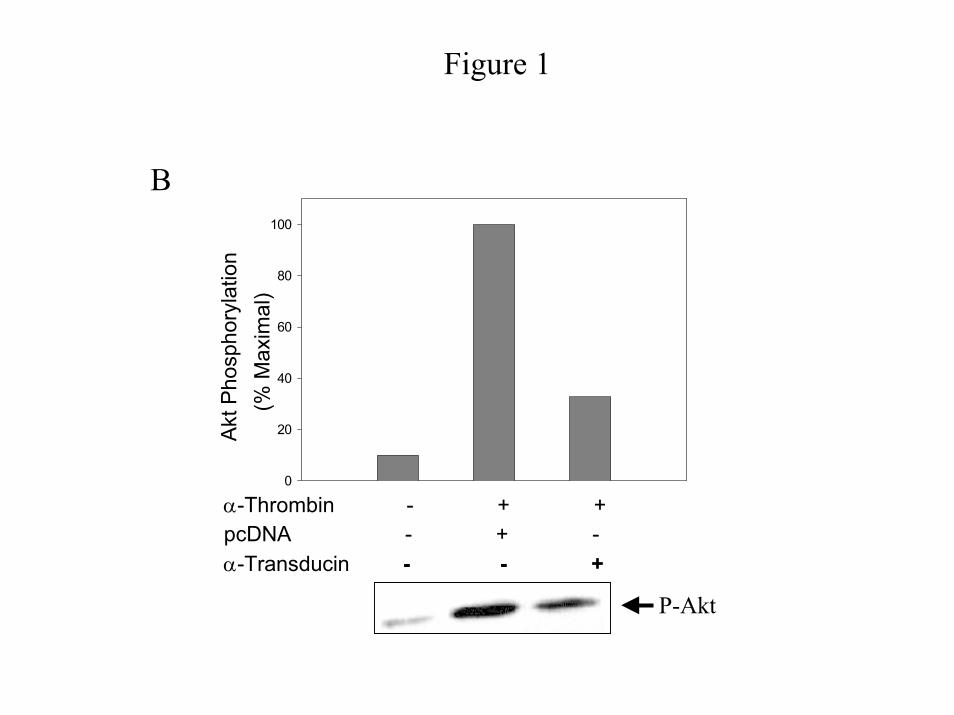
Figure 1
0
20
40
60
80
100A
kt P
hosp
hory
latio
n (%
Max
imal
)
α-Thrombin - + + pcDNA - + -α-Transducin - - +
P-Akt
B
by guest on December 25, 2019 http://www.jbc.org/ Downloaded from
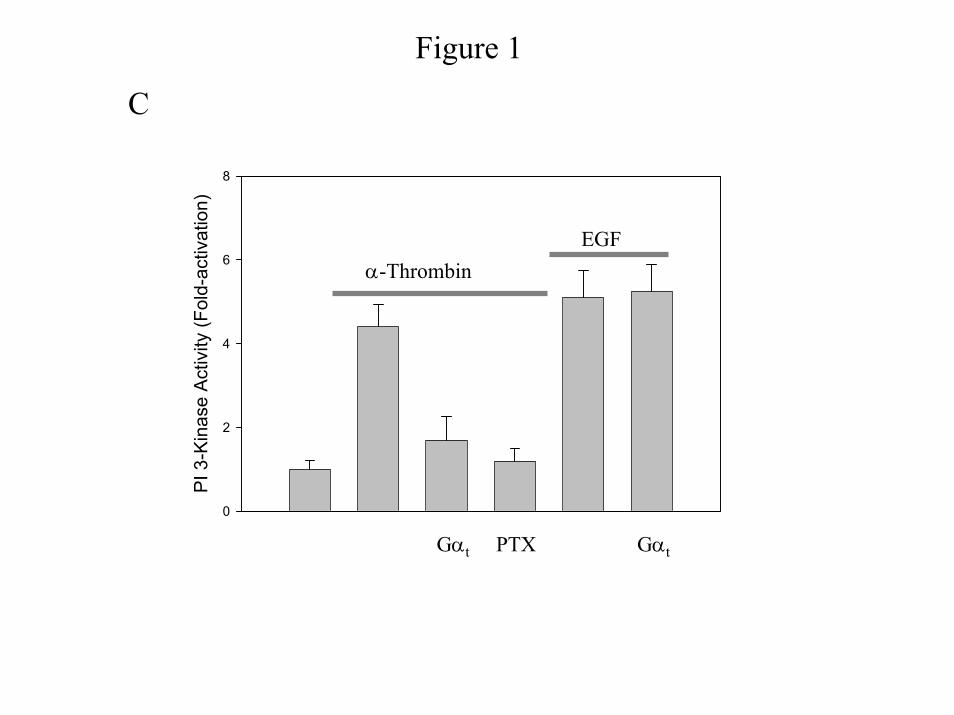
Figure 1
C
PI 3
-Kin
ase
Act
ivity
(Fol
d-ac
tivat
ion)
0
2
4
6
8
EGFα-Thrombin
Gαt PTX Gαt
by guest on December 25, 2019 http://www.jbc.org/ Downloaded from

0
1000
2000
3000
4000
5000
6000
7000
Gs Gi2 Gi3 Go Gq G13control
Figure 2AA
Imm
unop
rec i
pita
ted
[35S]
GT
P γS
( cpm
)
by guest on December 25, 2019 http://www.jbc.org/ Downloaded from

Figure 2
BB
0
2000
4000
6000
8000
Control Gi2 Gi3 Go
Imm
uno p
reci
pita
ted
[35S]
GTP
γ S (c
pm)
by guest on December 25, 2019 http://www.jbc.org/ Downloaded from
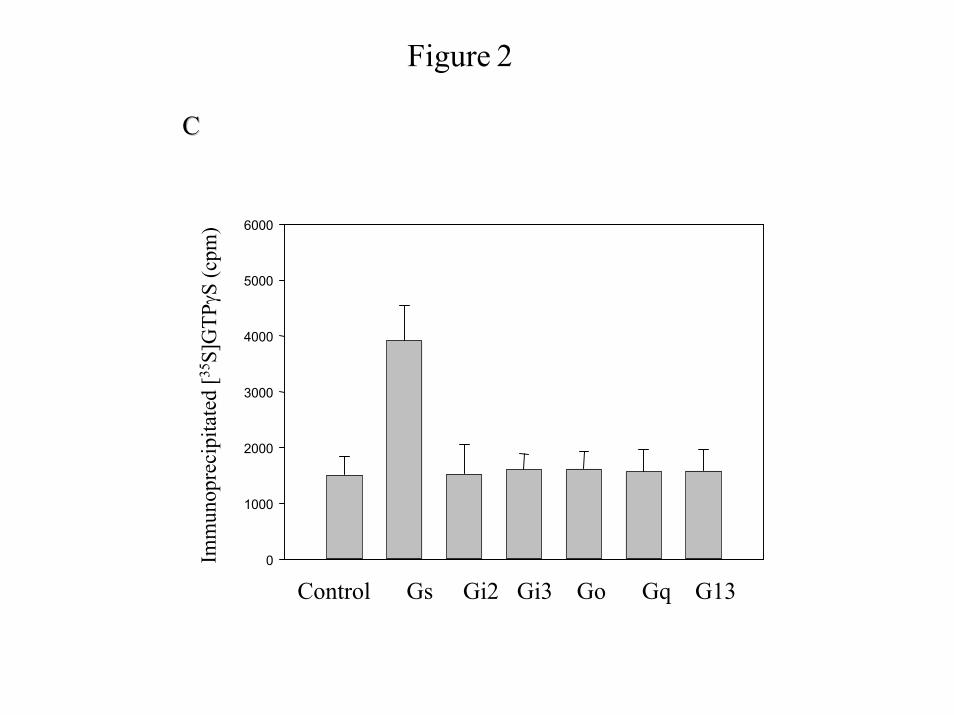
Figure 2
0
1000
2000
3000
4000
5000
6000
Control Gs Gi2 Gi3 Go Gq G13
CC
Imm
uno p
reci
pita
ted
[35S]
GTP
γ S (c
pm)
by guest on December 25, 2019 http://www.jbc.org/ Downloaded from
0
5
10
15
20
25
30
PTX - - + - - - - - -C terminal peptide Gαi/2 Gαq Gα13
GαXs Gα13 Gαi2 Gαq
Akt
Pho
spho
ryla
tion
(% M
ax)
α-Thrombinα-Thrombin
Figure 3A
P-Akt
Akt
by guest on December 25, 2019 http://www.jbc.org/ Downloaded from
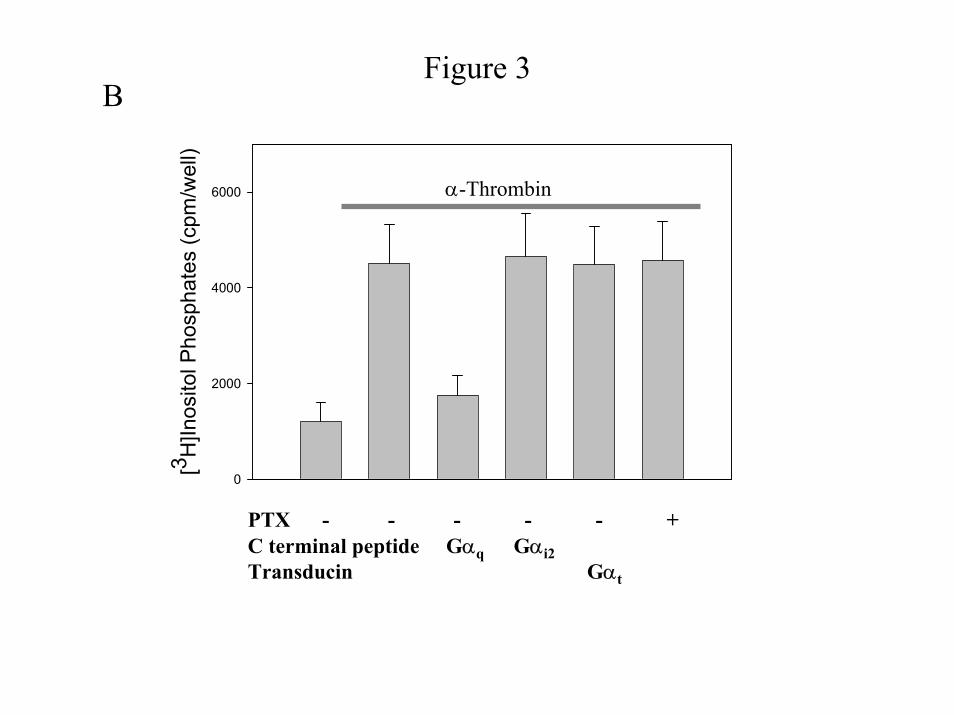
Figure 3B
PTX - - - - - + C terminal peptide Gαq Gαi2 Transducin Gαt
[3 H]In
osito
l Pho
spha
tes
(cpm
/wel
l)
0
2000
4000
6000 α-Thrombin
by guest on December 25, 2019 http://www.jbc.org/ Downloaded from
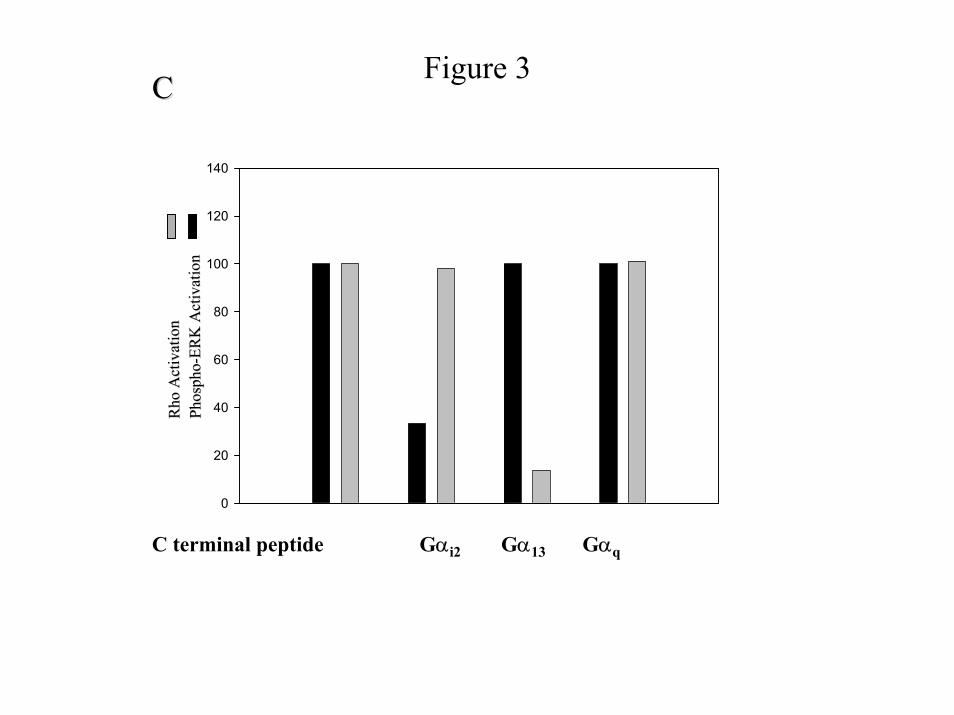
Figure 3CC
C terminal peptide Gαi2 Gα13 Gαq
0
20
40
60
80
100
120
140R
hoRho
Act
ivat
ion
Act
ivat
ion
Phos
pho
Phos
pho --
ERK
Act
ivat
ion
ERK
Act
ivat
ion
by guest on December 25, 2019 http://www.jbc.org/ Downloaded from
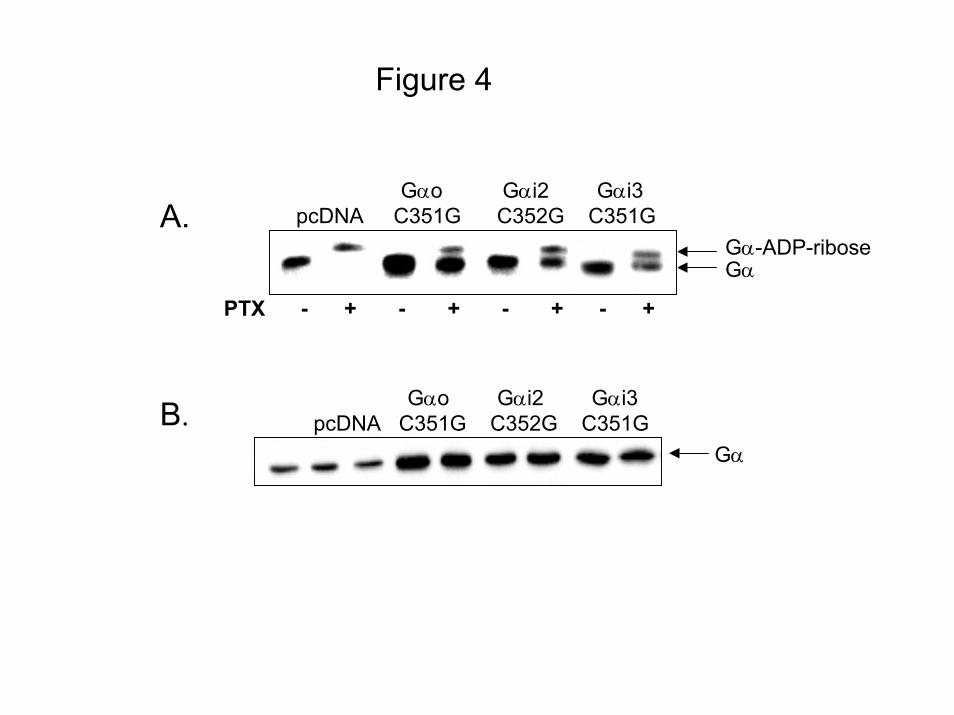
Figure 4
Gα-ADP-riboseGα
PTX - + - + - + - +
Gαo Gαi2 Gαi3pcDNA C351G C352G C351GA.
Gαo Gαi2 Gαi3pcDNA C351G C352G C351G
Gα
B.
by guest on December 25, 2019 http://www.jbc.org/ Downloaded from
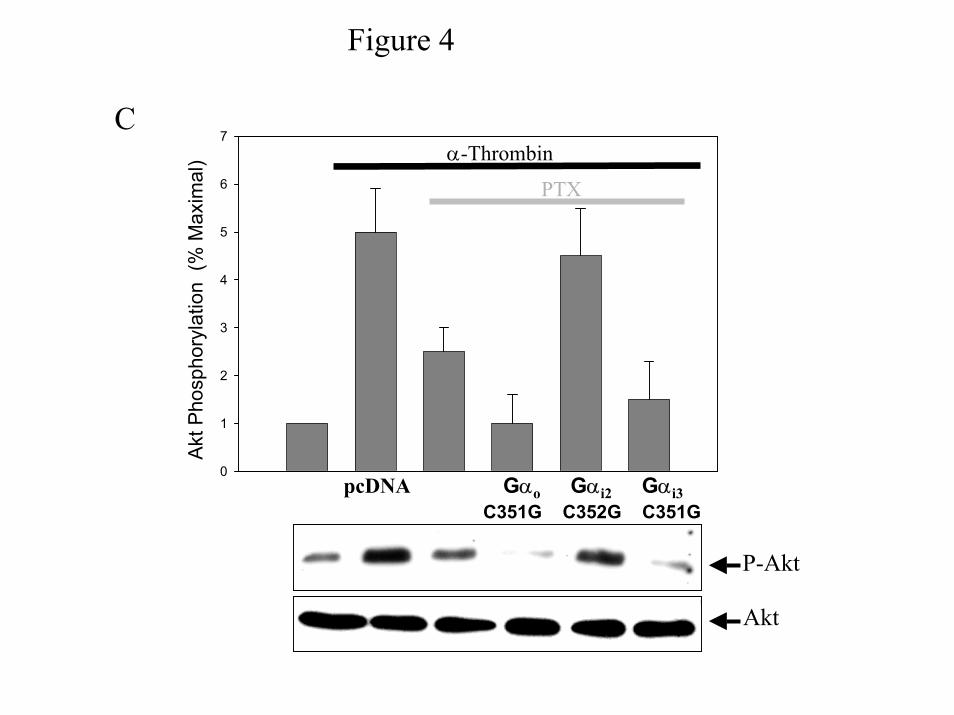
Figure 4
0
1
2
3
4
5
6
7
Akt
Pho
spho
ryla
tion
(% M
axim
al)
pcDNA Gαo Gαi2 Gαi3C351G C352G C351G
P-Akt
Akt
α-Thrombin
PTX
C
by guest on December 25, 2019 http://www.jbc.org/ Downloaded from
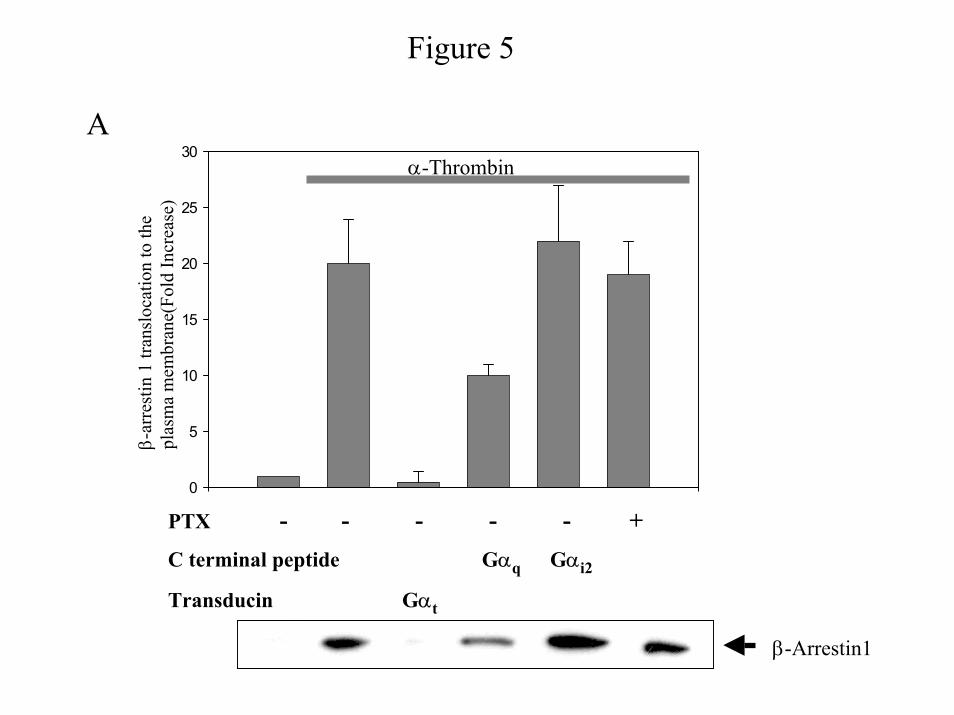
Figure 5
0
5
10
15
20
25
30α-Thrombin
β-ar
rest
in 1
tran
sloc
atio
n to
the
plas
ma
mem
bran
e(Fo
ld In
crea
se)
A
PTX - - - - - +C terminal peptide Gαq Gαi2
Transducin Gαt
β-Arrestin1
by guest on December 25, 2019 http://www.jbc.org/ Downloaded from
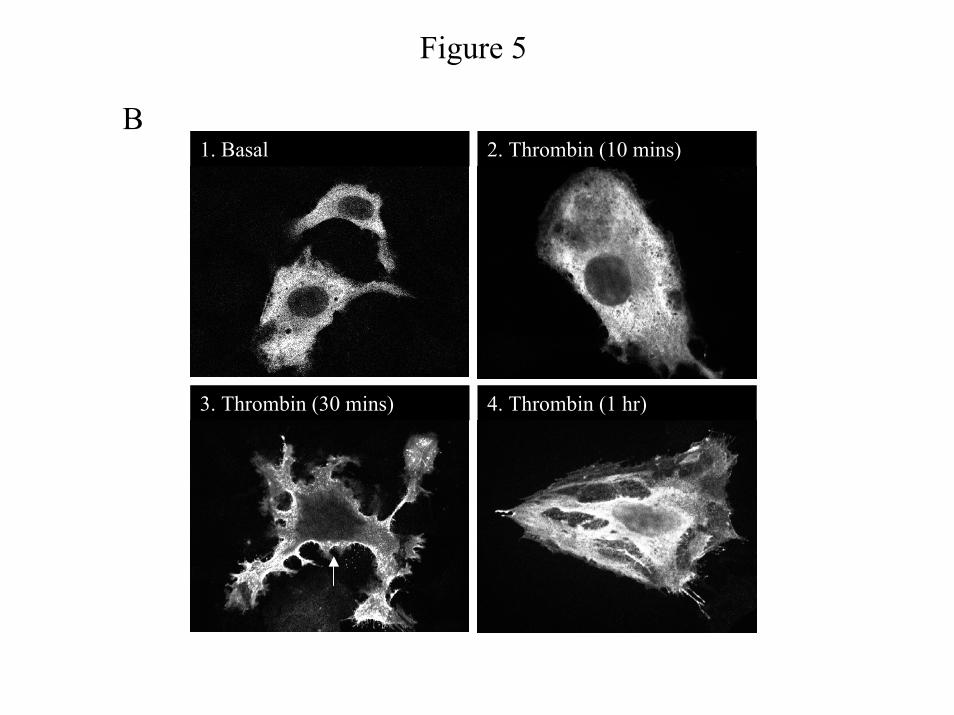
Figure 5
B1. Basal 2. Thrombin (10 mins)
3. Thrombin (30 mins) 4. Thrombin (1 hr)
by guest on December 25, 2019 http://www.jbc.org/ Downloaded from

Figure 5C
5. α-Thrombin + Gαt 6. α-Thrombin + C-terminal Gi1/2
1. Unstimulated
3. α-Thrombin + PTX
2. α-Thrombin
4. α-Thrombin + C-terminal Gq
by guest on December 25, 2019 http://www.jbc.org/ Downloaded from
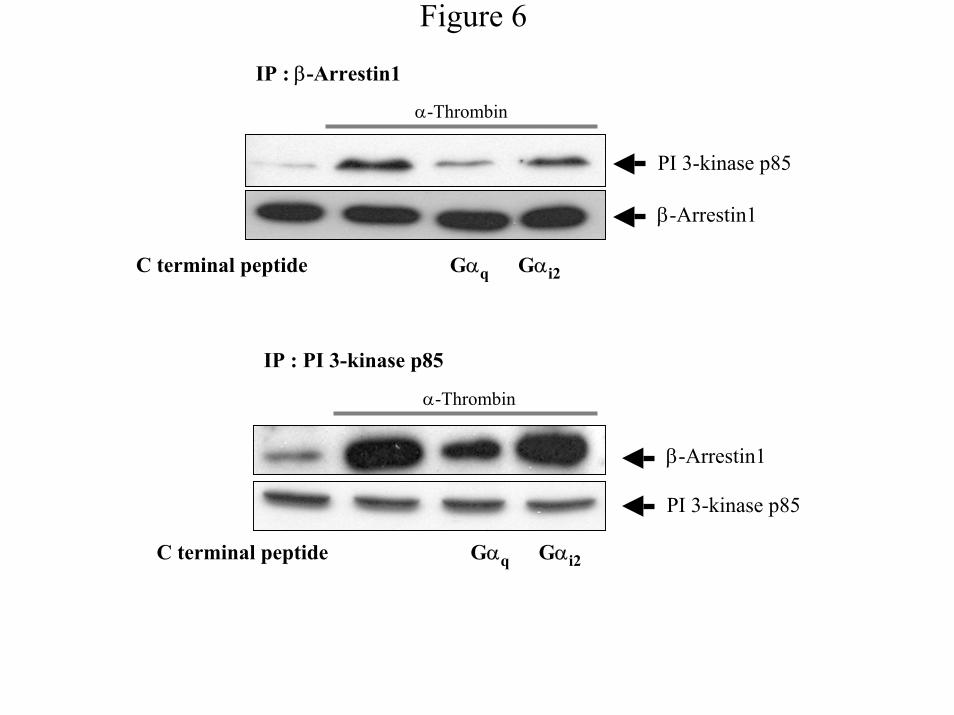
Figure 6
IP : β-Arrestin1
α-Thrombin
IP : PI 3-kinase p85
β-Arrestin1
PI 3-kinase p85
β-Arrestin1
PI 3-kinase p85
C terminal peptide Gαq Gαi2
α-Thrombin
C terminal peptide Gαq Gαi2
by guest on December 25, 2019 http://www.jbc.org/ Downloaded from
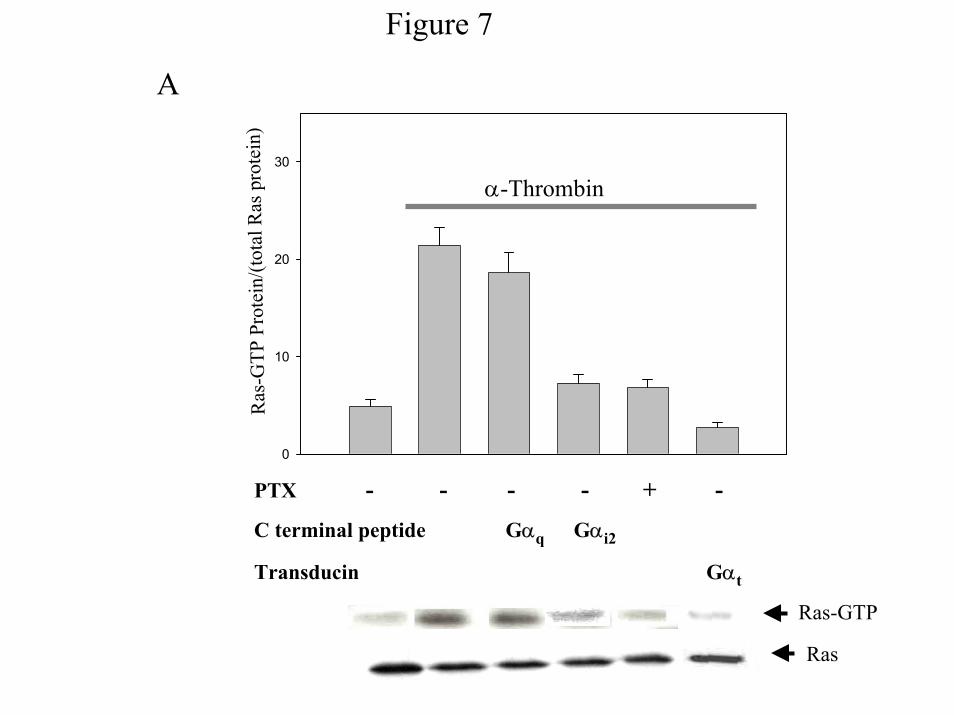
Figure 7
A
0
10
20
30
Ras-GTP
PTX - - - - + -
C terminal peptide Gαq Gαi2
Transducin Gαt
Ras
α-Thrombin
Ras
-GTP
Pro
tein
/(tot
al R
as p
rote
in)
by guest on December 25, 2019 http://www.jbc.org/ Downloaded from

Figure 7
0
5
10
15
20
25
30
35
pcDNA Gαo Gαi2 Gαi3(C351G) (C352G) (C351G)
PTX - - + - + - + - +
α-Thrombin
Ras-GTP
Ras
B
Ras
-GTP
Pro
tein
/(tot
al R
as p
rote
in)
by guest on December 25, 2019 http://www.jbc.org/ Downloaded from
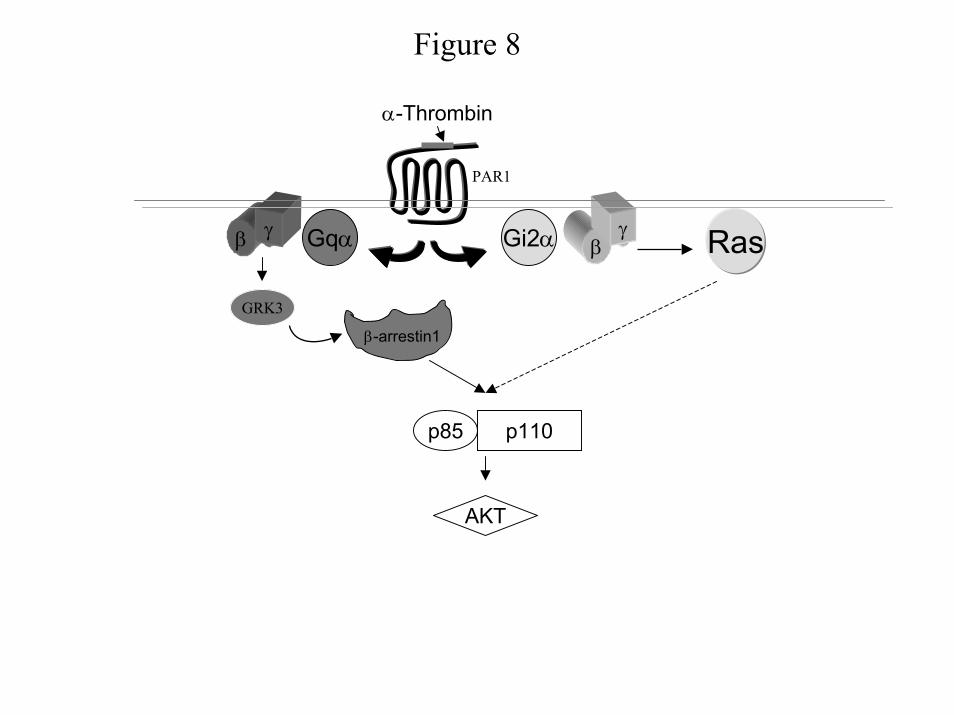
Figure 8
RasβγGi2α
p85 p110
Gqαβ γ
GRK3
β-arrestin1
α-Thrombin
PAR1
AKT
by guest on December 25, 2019 http://www.jbc.org/ Downloaded from

BaldassareReema Goel, Polly J. Phillips-Mason, Alice Gardner, Daniel M. Raben and Joseph J.
scripti2αscriptq and Gα dimers from Gγβ-Thrombin mediated PI 3-kinase activation through release of Gα
published online December 9, 2003J. Biol. Chem.
10.1074/jbc.M308753200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
by guest on Decem
ber 25, 2019http://w
ww
.jbc.org/D
ownloaded from


















