
- Cloud Object Storage | Store & … Spicer, MD, Richard Sposto, PhD, and Debu Tripathy, MD MelanoMa...
56
The T H E O F F I C I A L J O U R N A L O F of LUNG CANCER Lung Cancer Disparities in the Era of Personalized Medicine Christopher S. Lathan, MD, MS, MPH LEUKEMIA Expert Perspective on ASH 2014: Leukemia Meir Wetzler, MD, FACP PROSTATE CANCER Anti-Androgen Therapies for Prostate Cancer: A Focused Review Nischala Ammannagari MD, and Saby George MD, FACP BREAST CANCER Efficacy of Very-Low-Dose Capecitabine in Metastatic Breast Cancer Caitlin Bertelsen, MD, Lingyun Ji, MS, Agustin A. Garcia, MD, Christy Russell, MD, Darcy Spicer, MD, Richard Sposto, PhD, and Debu Tripathy, MD MELANOMA SPECIAL SECTION COMMENTARY New Year’s Resolution: Work to Do Omid Hamid, MD The Evolving Role of Surgery in Advanced Melanoma Richard Essner, MD, FACS PERSPECTIVE Is There an Optimal Intersection for Targeted and Immunotherapy Treatments for Melanoma? Jason J. Luke, MD, FACP Radiation and Melanoma: A Phoenix Rising Stephen L. Shiao, MD, PhD, and Omid Hamid, MD American Journal Hematology/ Oncology ® A Peer-Reviewed Resource for Oncology Education ajho www.AJHO.com ISSN 1939-6163 (print) ISSN 2334-0274 (online) Volume 11 Number 2 2.15 BREAST CANCER CME-certified enduring materials sponsored by Physicians’ Education Resource ® , LLC Recap of SABCS 2014: Changes for Today and Hope for Tomorrow
Transcript of - Cloud Object Storage | Store & … Spicer, MD, Richard Sposto, PhD, and Debu Tripathy, MD MelanoMa...

T h e
TH
E O
FFICIAL J
OU
RN
AL O
F o f
Lung CanCer Lung Cancer Disparities in the Era of Personalized MedicineChristopher S. Lathan, MD, MS, MPH
Leukemia Expert Perspective on ASH 2014: LeukemiaMeir Wetzler, MD, FACP
Prostate CanCerAnti-Androgen Therapies for Prostate Cancer: A Focused ReviewNischala Ammannagari MD, and Saby George MD, FACP
Breast CanCerEfficacy of Very-Low-Dose Capecitabine in Metastatic Breast CancerCaitlin Bertelsen, MD, Lingyun Ji, MS, Agustin A. Garcia, MD, Christy Russell, MD, Darcy Spicer, MD, Richard Sposto, PhD, and Debu Tripathy, MD
MelanoMa Special Section
CommentaryNew Year’s Resolution: Work to DoOmid Hamid, MD
The Evolving Role of Surgery in Advanced MelanomaRichard Essner, MD, FACS
PersPeCtiveIs There an Optimal Intersection for Targeted and Immunotherapy Treatments for Melanoma?Jason J. Luke, MD, FACP
Radiation and Melanoma: A Phoenix RisingStephen L. Shiao, MD, PhD, and Omid Hamid, MD
A m e r i c a n
J o u r n a l
H e m a t o l o g y /
O n c o l o g y ®
a Peer-reviewed resource
for oncology education
ajho
www.AJHO.com issn 1939-6163 (print) issn 2334-0274 (online)
Volume 11 Number 2 2.15
Breast CanCer CME-certified enduring materials sponsored by Physicians’ Education Resource®, LLC
Recap of SABCS 2014: Changes for Today and Hope for Tomorrow

Physicians Education Resource,® LLC Advancing Cancer Care Through Professional Education®
MAPPED!®The 30 Second Guide to the New GoToPER.com
Brought to you by
Online CME ActivitiesQuickly find Online Activities that discuss issues that you are seeing in your practice today.
American Journal of Hematology/OncologyStay up to date with news and recent research with the official journal of PER®.
Executive BoardMeet the PER® Executive Board – our advisory board of the top cancer experts.
Customize Your CME CalendarBe sure not to miss the CME Activities that you have registered for.
Manage Your CMEKeep track of what CME activities you have participated in, request credit, and receive certificates of completion.
Live CME ActivitiesEasily view and register to join our colleagues at one of our upcoming best-in-class CME conferences.
Navigate by Tumor TypeQuickly find live and online CME-certified activities related to your interests.
Take GoToPER.com On-the-GoGoToPER.com is mobile friendly. Easily view all that PER® has to offer on your hand-held mobile device or tablet, anytime, anywhere!
Breast Cancer
DermatologicCancer
GastrointestinalCancer
GenitourinaryCancer
GynecologicCancer
Head & NeckCancer
HematologicMalignancies
Lung Cancer
Prostate Cancer
Targeted Therapies
Share With Your ColleaguesQuickly share links to your favorite CME-certified activities with our social media buttons.
Mapped! is a registered trademark of Michael J. Hennessy Associates, Inc.
Plainsboro, NJ 08536
Physicians’ Education Resource®, LLC is accredited by the Accreditation Council for Continuing Medical Education to provide continuing medical education for physicians.
PER-Website-2015-Mapped_Asize.indd All Pages 1/30/15 10:37 AM

Physicians Education Resource,® LLC Advancing Cancer Care Through Professional Education®
MAPPED!®The 30 Second Guide to the New GoToPER.com
Brought to you by
Online CME ActivitiesQuickly find Online Activities that discuss issues that you are seeing in your practice today.
American Journal of Hematology/OncologyStay up to date with news and recent research with the official journal of PER®.
Executive BoardMeet the PER® Executive Board – our advisory board of the top cancer experts.
Customize Your CME CalendarBe sure not to miss the CME Activities that you have registered for.
Manage Your CMEKeep track of what CME activities you have participated in, request credit, and receive certificates of completion.
Live CME ActivitiesEasily view and register to join our colleagues at one of our upcoming best-in-class CME conferences.
Navigate by Tumor TypeQuickly find live and online CME-certified activities related to your interests.
Take GoToPER.com On-the-GoGoToPER.com is mobile friendly. Easily view all that PER® has to offer on your hand-held mobile device or tablet, anytime, anywhere!
Breast Cancer
DermatologicCancer
GastrointestinalCancer
GenitourinaryCancer
GynecologicCancer
Head & NeckCancer
HematologicMalignancies
Lung Cancer
Prostate Cancer
Targeted Therapies
Share With Your ColleaguesQuickly share links to your favorite CME-certified activities with our social media buttons.
Mapped! is a registered trademark of Michael J. Hennessy Associates, Inc.
Plainsboro, NJ 08536
Physicians’ Education Resource®, LLC is accredited by the Accreditation Council for Continuing Medical Education to provide continuing medical education for physicians.
PER-Website-2015-Mapped_Asize.indd All Pages 1/30/15 10:37 AM

Table of Contents
Lung CanCer
Lung Cancer Disparities in the Era of Personalized Medicine Christopher S. Lathan, MD, MS, MPHWidespread disparities by race and socioeconomic status in cancer outcomes are well documented. In lung cancer, black patients are less likely than white patients to receive stage-appropriate cancer care, including surgery, radiation, and systemic therapy. Reasons for these disparities are multifactorial and are discussed here.
Leukemia
Expert Perspective on ASH 2014: LeukemiaMeir Wetzler, MD, FACPAbstracts presented at the 2014 Annual Meeting and Exposition of the American Society of Hema-tology (ASH) signaled evolutions in treatment, as heralded by the promise of exciting new agents in development. This review presents highlights of key abstracts in leukemia.
Prostate CanCer
Anti-Androgen Therapies for Prostate Cancer: A Focused Review Nischala Ammannagari MD, and Saby George MD, FACPAndrogen deprivation is the mainstay of treatment in prostate cancer, and an increased understand-ing of the androgen receptor signaling pathway and mechanisms of resistance to castration over the past decade has led to the discovery of newer agents. In this article, the authors review novel targeted therapies in castration-resistant prostate cancer and biomarkers of resistance to such therapies.
Breast CanCer
Efficacy of Very-Low-Dose Capecitabine in Metastatic Breast Cancer Caitlin Bertelsen, MD, Lingyun Ji, MS, Agustin A. Garcia, MD, Christy Russell, MD, Darcy Spicer, MD, Richard Sposto, PhD, and Debu Tripathy, MDThe FDA-approved dose of capecitabine is often associated with treatment-limiting toxicities. Clinical experience and published reports suggest that lower starting doses of capecitabine can be as effective as the approved dose. In this retrospective analysis the authors compare the efficacy of significantly lower doses of capecitabine with the FDA-approved dose, using previously published results as com-parators.
Commentary
New Year’s Resolution: Work to DoOmid Hamid, MD As 2015 begins, it is with great anticipation that we await the results of ongoing work in the field of melanoma therapy. We have come a long way, as immunotherapy with PD-1/PD-L1 checkpoint inhibition has gained approval as a standard therapy. We have just begun to touch on the promise of immunotherapy. The future will elucidate optimal combinations of checkpoint inhibition and other immune-oncologic modalities and targeted agents.
The Evolving Role of Surgery in Advanced Melanoma Richard Essner, MD, FACS The rapid approval in recent years of several new therapies for melanoma have offered the hope of better outcomes, but the role of surgery in the treatment of advanced melanoma—in light of these newly available therapies—remains to be elucidated.
MelanoMa Special Section
5
9
15
20
31
33

Table of Contents (continued) PersPeCtive
Is There an Optimal Intersection for Targeted and Immunotherapy Treatments for Melanoma?Jason J. Luke, MD, FACPMelanoma therapeutics have undergone massive changes with the approval of BRAF and MEK kinase inhibi-tors and immune-checkpoint blocking antibodies against CTLA-4 and PD-1. Targeted and immunotherapy have different strengths and weaknesses, but both are essential to clinical management of patients with advanced melanoma.
Radiation and Melanoma: A Phoenix RisingStephen L. Shiao, MD, PhD, and Omid Hamid, MDMany oncologists do not consider radiation as a treatment option for melanoma, mainly due to the outdated perception that melanomas are “resistant” to radiation. Fortunately, the advent of new modalities of highly focused radiation therapy, the increasing understanding of the role of the immune system in regulating the response to radiation therapy, and the recent development of a multitude of immune-oncologic treatment mo-dalities should change the role of radiation therapy in the treatment of melanoma.
Cme
CME-certified enduring materials sponsored by Physicians’ Education Resource®, LLCBreast CanCer
Recap of SABCS 2014: Changes for Today and Hope for Tomorrow At the 2014 San Antonio Breast Cancer Symposium (SABCS), key developments included practice-changing data for the treatment of premenopausal breast cancer, early evaluation of immunotherapy in the treatment of triple negative breast cancer, outcomes from the ICE trial concerning the treatment of elderly patients with early-stage breast cancer, and the first presentation of a checkpoint inhibitor in breast cancer. This CME activity reviews select abstracts from SABCS 2014, chosen for their impact on current clinical practice or because they lay the groundwork for further investigations.
34
39
42
Patrick I. Borgen, MDChairman, Department of Surgery Maimonides Medical CenterDirector, Brooklyn Breast Cancer ProgramBrooklyn, NY
Julie R. Brahmer, MDAssociate Professor, Oncology Johns Hopkins University School of
MedicineSidney Kimmel Comprehensive Cancer
CenterBaltimore, MD
Myron S. Czuczman, MDProfessor of OncologyChief, Lymphoma/Myeloma ServiceDepartment of MedicineHead, Lymphoma Translational Research
LaboratoryDepartment of ImmunologyRoswell Park Cancer InstituteBuffalo, NY
David R. Gandara, MDProfessor of MedicineDirector, Thoracic Oncology ProgramSenior Advisor to the DirectorDivision of Hematology/Oncology
UC Davis Comprehensive Cancer Center Sacramento, CA
Andre Goy, MD, MSChairman and Director Chief of LymphomaDirector, Clinical and Translational Cancer ResearchJohn Theurer Cancer Center at Hackensack University Medical CenterHackensack, NJ
John M. Kirkwood, MDUsher Professor of Medicine, Dermatology
and Translational ScienceDirector, Melanoma and Skin Cancer
ProgramUPMC Hillman Cancer CenterPittsburgh, PA
Michael Kolodziej, MD National Medical Director, Oncology Solutions
Office of the Chief Medical Officer, AetnaHartford, CT
Maurie Markman, MDPresident, Medicine & ScienceNational Director, Medical OncologyCancer Treatment Centers of America
John L. Marshall, MDChief, Hematology and Oncology Director, Otto J. Ruesch Center for the
Cure of Gastrointestinal CancersLombardi Comprehensive Cancer CenterGeorgetown University Medical CenterWashington, DC
Joyce A. O’Shaughnessy, MDCo-Director, Breast Cancer ResearchBaylor Charles A. Sammons Cancer
Center Texas Oncology The US Oncology NetworkDallas, TX
Daniel P. Petrylak, MDProfessor of Medicine (Medical Oncology) and of UrologyCo-Director, Signal Transduction Research
ProgramYale Cancer Center and Smilow Cancer
HospitalNew Haven, CT
Ramesh K. Ramanathan, MDProgram Lead, Gastrointestinal OncologySenior Associate AttendingMayo Clinic, ArizonaClinical Professor, Translational Genomics
Research Institute (TGEN)Phoenix, AZ
PER® Executive Board/AJHO Editorial Board

Editor-in-ChiefDebu Tripathy, MD
Professor and Chair Department of Breast Medical Oncology The University of Texas MD Anderson Cancer Center Houston, TX
Associate EditorMyron S. Czuczman, MD
Professor of OncologyChief, Lymphoma/Myeloma ServiceHead, Lymphoma Translational Research LaboratoryDepartment of ImmunologyRoswell Park Cancer InstituteBuffalo, NY
Managing EditorDevera Pine [email protected]
Editorial OfficesPhysicians’ Education Resource®, LLC666 Plainsboro Road, Ste. 356Plainsboro, NJ 08536(609) 378-3701
Medical DirectorMichael Perlmutter, MS, PharmD
eDitoriaL staFFFrom the Editor
This month’s issue of AJHO provides a deep dive into several areas of contemporary oncology practice.
Advances in melanoma are occurring on several fronts. After decades of little progress, durable responses
are being seen with a panoply of targeted therapies. Accordingly, this field has accounted for numerous
articles in the journal over the past few months. This issue brings a full collection of pieces on melano-
ma addressing surgery, radiation, and several targeted medical therapies, along with commentaries that
provide insights into controversies and emerging new standards.
A review on the new generation of androgen receptor pathway inhibitors presented by Drs Amman-
nagari and George summarizes recent advances in hormonal therapies that demonstrated activity after
chemotherapy, with ongoing trials that may reorder the sequence in which we use these drugs. A lung
cancer update from Dr Lathan addresses the clinical consequences of disparities in both patient care and
access, as well as biological differences among ethnicities and other population categories.
In the area of breast cancer, an original study questions the FDA-approved dosing of capecitabine, a
drug that is most commonly used at lower dosages with similar outcomes. However, formal demonstra-
tion of equivalent efficacy of dosages used in the community is lacking and has led to wide variations in
care patterns.
AJHO presents the second in a series of 56th American Society of Hematology (ASH) Annual Meeting
highlights, this one from Dr Wetzler, focusing on leukemia advances with a range of new agents including
sorafenib, IDH2-inhibiting AG-221, and FLT3-inhibiting quizartinib, as well as very exciting immuno-
logical engineered approaches using chimeric antigen receptor (CAR)-modified T-cells and the bispecific
T-cell engager (BiTE) antibody blinatumomab.
The CME article this month reviews key abstracts presented at the 2014 San Antonio Breast Cancer
Symposium that are potentially practice-changing, opening new research avenues. Specifically, will ovarian
blockade become a new standard for premenopausal patients? Is immunotherapy coming of age as the
first active nonchemotherapeutic approach demonstrated in triple-negative breast cancer?
As always, we welcome your comments.
Debu Tripathy, MD Editor-in-Chief
CorPorate oFFiCers Chairman and CEOMike Hennessy
Vice Chairman Jack Lepping
President Tighe Blazier
Chief Financial Officer Neil Glasser, CPA/CFE
Executive Vice President and General Manager John Maglione
Senior Vice President, Operations and Clinical Affairs Jeff Prescott, PharmD, RPh
Vice President, Human Resources Rich Weisman
Vice President, Executive Creative Director Jeff Brown
the content of this publication is for general information purposes only. the reader is encouraged to confirm the information presented with other sources. American Journal of Hematology/Oncology makes no representations or warranties of any kind about the completeness, accuracy, timeliness, reliability, or suitability of any of the information, including content or advertise-ments, contained in this publication and expressly disclaims liability for any errors and omissions that may be presented in this publication. American Journal of Hematology/Oncology re-serves the right to alter or correct any error or omission in the information it provides in this publication, without any obligations. American Journal of Hematology/Oncology further disclaims any and all liability for any direct, indirect, consequential, special, exemplary, or other damages arising from the use or misuse of any material or information presented in this publication. the views expressed in this publication are those of the authors and do not necessarily reflect the opinion or policy of American Journal of Hematology/Oncology.

VOL. 11, NO. 2 THE AMERICAN JOURNAL OF HEMATOLOGY/ONCOLOGY 5
· lung cancer ·
Lung Cancer Disparities in the Era of Personalized Medicine
Christopher S. Lathan, MD, MS, MPH
IntroductionWidespread disparities in cancer morbidity and mortality by race and socioeconomic status are well documented. These dispari-ties exist throughout all areas of the cancer spectrum, spanning screening, diagnosis, and treatment, as well as survivorship and end-of-life care. They are seen in multiple cancer types and af-fect both genders.1-4 The causes of disparities are multifactorial, encompassing access to care, neighborhood/residential factors, and patient and provider factors. Some studies have focused on trust issues, especially for African Americans, who, it is hypothe-sized, may perceive the health establishment in a more negative light given past mistreatment, and therefore refuse care at a high-er rate.5-7
While the preponderance of the published work in disparities discusses African Americans, other studies have reported dispar-ities in other racial and ethnic minorities and in patients with lower socioeconomic status.8-11 Due to a multitude of factors, patients with the most need have the greatest difficulty accessing high-level tertiary center cancer care.12-14 Healthcare systems that provide universal access have been shown to attenuate racial and ethnic disparities in treatment, evidence that further supports the important role of income and access in explaining observed
differences by race and class.15,16 Access to high-quality care has particular resonance in the age of personalized medicine because it is no longer just a theoretical possibility, but rather a well-estab-lished mode of treatment.
Underserved Patients With Lung CancerOncology treatment has made advancements in personalized medicine in many areas, but the disease that best illustrates the potential challenges for underserved patients is lung cancer. Lung cancer is the leading cause of cancer mortality for both men and women in the United States, with estimates accounting for 159,480 deaths in the year 2013.17 African-American men have the highest incidence and mortality rate of lung cancer.18-23
Moreover, African-American patients, both male and female, are less likely than white patients to receive stage-appropriate cancer care, including surgery, radiation, and systemic therapy.12,21-23,25-30
Reasons for these disparities are multifactorial, with contribu-tions from patients, providers, disease-related factors, as well as the effects of residential segregation.18,31-33 Socioeconomic status is closely tied to tobacco addiction and to poorer outcomes in lung cancer.34,35 This creates a situation in which low income increases the risk of lung cancer and increases the risk of dying from lung cancer, presumably from lack of appropriate treat-ment.11,36
Personalized medicine and—specifically for lung cancer—tar-geted therapy, is relevant for a sizable portion of patients, and there has been little research on lung cancer disparities by race/ethnicity in treatment with the targeted agents.37-46 Frequencies of other genomically altered therapeutic targets (EGFR, BRAF, ALK, MET, ROS-1, ERBB2) are not well characterized in Af-rican-American populations to the same extent as they are in Asian and white populations, and there are even fewer in studies of patients who identify as Latino.44,47 Indeed, the scope of dif-fusion of this treatment approach beyond the research centers has not been well characterized. Lynch et al48 demonstrated a decrease in EGFR mutation testing as distance from National Cancer Institute cancer centers increased. This suggests that the diffusion of personalized medicine might not be making it out of the cancer center and into the communities where underserved
Abstract
Widespread disparities by race and socioeconomic status
in cancer outcomes are well documented. These dispar-
ities exist throughout all areas of the cancer spectrum,
spanning screening, diagnosis, and treatments, as well
as survivorship and end-of-life care. African-American
patients and other racial/ethnic minorities are less likely
than white patients to receive stage-appropriate cancer
care, including surgery, radiation, and systemic therapy.
The impact of personalized medicine on disparities is dis-
cussed here, along with a novel community-based inter-
vention to address these disparities.
Key words: lung cancer, disparities, ethnicity, personal-
ized medicine, epidemiology, vulnerable populations

6 www.ajho.com FEbRUARY 2015
· lung cancer ·
patients reside.The recruitment of vulnerable patient populations into clini-
cal trials has also been challenging.49 Approximately 3% to 5% of all adult patients with cancer are on clinical trials, and while there remains some debate about representation by race, the to-tal number of adult patients of color or low socioeconomic sta-tus remains small.50,51 The reason for low enrollment of underrep-resented patients (defined as lower socioeconomic status, elderly, and racial/ethnic minorities) is likely due to a combination of factors including decreased access to clinical trials and physician triage approaches.50 As noted by Ford et al,51 “this lack of diver-sity in randomized study populations reduces opportunities for discovering effects that may be particularly relevant to underrep-resented populations.” The lack of data on molecular targets in lung cancer for African Americans is a specific example of this problem.52
An Intervention for Improving Outcomes for AllAs personalized medicine becomes standard, there is a possibil-ity that cancer treatment outcomes could worsen for underrep-resented populations, even as treatments improve for the gen-eral population.53 It is our hypothesis that increasing access to high-quality cancer care, improving relationships, and providing education within the community will remove some of the struc-tural barriers to clinical trial enrollment.
It is with this in mind that we developed an intervention to address the issues of access to high-level cancer care. The details of the intervention are published elsewhere.54,55 The goal of the intervention is to improve local oncology outcomes for the un-derserved by facilitating clinical access to preventive medicine, treatment, and clinical trials. The program provides on-site eval-uation services by oncologists to vulnerable populations in their community clinics in coordination with their primary care pro-viders, and expedited referrals to the cancer center for patients with an active cancer-related issue.
The intervention is based on a nurse navigation model, with an increased presence of medical oncology clinicians in the community health center setting. Patients are evaluated for the entire spectrum of oncology-related issues, including screening, diagnosis, and survivorship. Acute treatment for malignan-cy is performed at the cancer center. Certainly, the treatment advances that are developed in research labs, great and small, should be made accessible to the communities most affected by cancer. Far too often, cutting-edge treatment approaches are available solely to those who have the means to obtain them. In order to combat disparities by race/ethnicity and income, more community-based interventions are needed to determine what approaches will increase access to high-level care and improve
outcomes for all cancer patients. Affiliation: Christopher S. Lathan, MD, MS, MPH, is assistant professor of Medicine at Harvard Medical School, and faculty director for the Cancer Care Equity Program at the Dana-Farber Cancer Institute, boston, MA.Disclosure: Dr Lathan reports no relevant conflicts of interest to disclose. Address correspondence to: Christopher S. Lathan, MD, MS, MPH, 450 brookline Ave, D1120, boston, MA 02215. Email: [email protected].
RefeRenCes1. balboni TA, Vanderwerker LC, block SD, et al. Religiousness and spiritual support among advanced cancer patients and associations with end-of-life treatment preferences and quality of life. J Clin Oncol. 2007;25:555-560.2. Wright AA, Keating NL, balboni TA, et al. Place of death: correlations with quality of life of patients with cancer and predictors of bereaved caregivers’ mental health. J Clin Oncol. 2010;28:4457-4464.3. Wright AA, Mack JW, Kritek PA, et al. Influence of patients’ preferences and treatment site on cancer patients’ end-of-life care. Cancer. 2010;116:4656-4663.4. Potosky AL Saxman S, Wallace Rb, Lynch CF. Population variations in the initial treatment of non-small cell lung cancer. J Clin Oncol. 2004;22:3261-3268.5. Kagawa-Singer M, Dadia AV, Yu MC, et al. Cancer, culture, and health disparities: time to chart a new course? CA Cancer J Clin. 2010;60:12-39.6. Niu X, Pawlish KS, Roche LM. Cancer survival disparities by race/ethnicity and socioeconomic status in New Jersey. J Health Care Poor Underserved. 2010;21:144-160.7. Onega T, Duell EJ, Shi X, et al. Race versus place of service in mortality among medicare beneficiaries with cancer. Cancer. 2010;116:2698-2706.8. Tian N, Goovaerts P, Zhan Fb, et al. Identifying risk factors for disparities in breast cancer mortality among African-American and Hispanic women. Womens Health Issues. 2013;22:e267-e276.9. Miranda PY, Tarraf W, Gonzalez HM. breast cancer screening and ethnicity in the United States: implications for health disparities research. Breast Cancer Res Treat. 2011;128:535-542.10. Gross CP, Filardo G, Mayne ST, et al. The impact of socioeconomic status and race on trial participation for older women with breast cancer. Cancer. 2005;103:483-491.11. Albano JD, Ward E, Jemal A, et al. Cancer mortality in the United States by education level and race. J Natl Cancer Inst. 2007;99:1384-1394.

VOL. 11, NO. 2 THE AMERICAN JOURNAL OF HEMATOLOGY/ONCOLOGY 7
Lung CanCer Disparities in the era of personaLizeD MeDiCine
12. Clegg LX, Reichman ME, Miller bA, et al. Impact of socioeconomic status on cancer incidence and stage at diagnosis: selected findings from the surveillance, epidemiology, and end results: National Longitudinal Mortality Study. Cancer Causes Control. 2009;20:417-435.13. American Cancer Society: Cancer facts and figures 2013. http://www.cancer.org/research/cancerfactsfigures/cancerfactsfigures/ cancer-facts-figures-2013. Accessed January 31, 2015.14. bradley CJ, Dahman b, Given CW. Inadequate access to surgeons: reason for disparate cancer care? Med Care. 2009;47:758-764.15. blackstock AW, Herndon JE, Paskett Ed, et al. Outcomes among African-American/non-African American patients with advanced non small cell lung carcinoma: report from the Cancer and Leukemia Group b. J Natl Cancer Inst. 2002;94:284-290.16. Mulligan CR, Meram AD, Proctor CD, et al. Unlimited access to care: effect on racial disparity and prognostic factors in lung cancer. Cancer Epidemiol Biomarkers Prev. 2006;15:25-31.17. American Cancer Society: Cancer facts and figures 2008. http://www.cancer.org/research/cancerfactsstatistics/cancerfactsfigures2008/index. Accessed January 31, 2015.18. Stewart J. Lung cancer in African Americans. Cancer. 2001;91:2476-2481.19. bach Pb CL, Warren JL, begg Cb. Racial differences in the treatment of early-stage lung cancer. N Engl J Med. 1999;34:1198-1205.20. bach Pb, Cramer LD, Schrag D, et al. The influence of hospital volume on survival after resection for lung cancer. N Engl J Med. 2001;345:181-188.21. Lathan CS, Neville bA, Earle CC. The effect of race on invasive staging and surgery in non-small-cell lung cancer. J Clin Oncol. 2006;24:413-418.22. Lathan CS, Neville bA, Earle CC. Racial composition of hospitals: effects on surgery for early-stage non-small-cell lung cancer. J Clin Oncol. 2008;26:4347-4352.23. Earle C, Venditti LN, Nuemann, PJ, et al. Who gets chemotherapy for metastatic lung cancer? Chest. 2000;117:1239-1246.24. Earle CC, Neumann PJ, Gelber RD, et al. Impact of referral patterns on the use of chemotherapy for lung cancer. J Clin Oncol. 2002;20:1786-1792.25. Cykert S, Phifer, N. Surgical decisions for early stage non-small cell lung cancer: which racially sensitive perceptions of cancer are likely to explain racial variation in surgery? Med Decis Making. 2003;23:167-176.26. DeLancey JO, Thun MJ, Jemal A, et al. Recent trends in black-White disparities in cancer mortality. Cancer Epidemiol Biomarkers Prev. 2008;17:2908-2912.
27. Esnaola NF, Gebregziabher M, Knott K, et al. Underuse of surgical resection for localized, non-small cell lung cancer among whites and African Americans in South Carolina. Ann Thorac Surg. 2008;86:220-226; discussion 227.28. Murphy MM, Tseng JF, Shah SA. Disparities in cancer care: an operative perspective. Surgery. 2010;147:733-737.29. Polite bN, Dignam JJ, Olopade OI. Colorectal cancer model of health disparities: understanding mortality differences in minority populations. J Clin Oncol. 2006;24:2179-2187.30. Akinyemiju TF, Soliman AS, Johnson NJ, et al. Individual and neighborhood socioeconomic status and healthcare resources in relation to black-White breast cancer survival disparities. J Cancer Epidemiol. 2013;2013:490472.31. Stellman S, Chen Y, Muscat JE, et al. Lung cancer risk in White and black Americans. Annals Epidemiol. 2003;13:294-302.32. Lathan C. Racial disparities in lung cancer. In: Kernstine KH, Reckamp KL (eds): Lung Cancer: A Multidisciplinary Approach to Diagnosis and Management. New York: Demos Medical Publishing, LLC; 2011:293-298.33. Institute of Medicine. Unequal Treatment. Washington, DC: The National Academies Press; 2003.34. Agaku IT, Vardavas CI, Ayo-Yusuf OA, et al. Gender and racial differences in smoking of long/ultra-long and king size cigarettes among US adult smokers, NHANES 1999-2012. Drug Alcohol Depend. 2014;136:28-35.35. businelle MS, Kendzor DE, Reitzel LR, et al. Mechanisms linking socioeconomic status to smoking cessation: a structural equation modeling approach. Health Psychol. 2010;29:262-273.36. Geyer S. Social inequalities in the incidence and case fatality of cancers of the lung, the stomach, the bowels, and the breast. Cancer Causes Control. 2008;19:965-974.37. Lynch TJ, bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129-2139.38. Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497-1500.39. Yang SH, Mechanic LE, Yang P, et al. Mutations in the tyrosine kinase domain of the epidermal growth factor receptor in non-small cell lung cancer. Clin Cancer Res. 2005;11:2106-2110.40. Krishnaswamy S, Kanteti R, Duke-Cohan JS, et al. Ethnic differences and functional analysis of MET mutations in lung cancer. Clin Cancer Res. 2009;15:5714-5723.41. Leidner RS, Fu P, Clifford b, et al. Genetic abnormalities of the EGFR pathway in African American Patients with non-small-cell lung cancer. J Clin Oncol. 2009;27:5620-5626.42. Cote ML, Haddad R, Edwards DJ, et al. Frequency and

8 www.ajho.com FEbRUARY 2015
· lung cancer ·
type of epidermal growth factor receptor mutations in African Americans with non-small cell lung cancer. J Thorac Oncol. 2011;6:627-630.43. Janku F, Garrido-Laguna I, Petruzelka Lb, et al. Novel therapeutic targets in non-small cell lung cancer. J Thorac Oncol. 2011;6:1601-1612.44. Mok TS. Personalized medicine in lung cancer: what we need to know. Nat Rev Clin Oncol. 2011;8:661-668.45. Paik PK, Arcila ME, Fara M, et al. Clinical characteristics of patients with lung adenocarcinomas harboring bRAF mutations. J Clin Oncol. 2011;29:2046-2051.46. Reinersman JM, Johnson ML, Riely GJ, et al. Frequency of EGFR and KRAS mutations in lung adenocarcinomas in African Americans. J Thorac Oncol. 2011;6:28-31.47. Mok TS, Lam KC. The diverse diversity. J Thorac Oncol. 2011;6:842-843.48. Lynch JA, Khoury MJ, borzecki A, et al. Utilization of epidermal growth factor receptor (EGFR) testing in the United States: a case study of T3 translational research. Genet Med. 2013;15:630-638.49. Joseph G, Dohan D. Recruiting minorities where they receive care: institutional barriers to cancer clinical trials recruitment in a safety-net hospital. Contemp Clin Trials. 2009;30:552-559.50. Colon-Otero G, Smallridge RC, Solberg LA, Jr, et al. Disparities in participation in cancer clinical trials in the United States: a symptom of a healthcare system in crisis. Cancer. 2008;112:447-454.51. Ford JG, Howerton MW, Lai GY, et al. barriers to recruiting underrepresented populations to cancer clinical trials: a systematic review. Cancer. 2008;112:228-242.52. Ma PC. Molecular genetic variations of lung cancer between human populations. Presented at: the American Society of Clinical Oncology; June 7, 2010; Chicago, IL. http://meetinglibrary.asco.org/content/39223?media=vm. Accessed January 31, 2015.53. Kaur JS, Petereit DG. Personalized medicine: challenge and promise. J Cancer Education. 2012;27:12-17.54. Waldman LT, Svoboda L, Young bF, et al. A novel community-based delivery model to combat cancer disparities. Healthcare. 2013;1:123-129.55. Waldman LT, bean W, Levine AL, et al. Using FastTrack to implement an academic medical center and community health center collaborative for cancer care delivery. Healthcare. 2013;1:130-135.

VOL. 11, NO. 2 THE AMERICAN JOURNAL OF HEMATOLOGY/ONCOLOGY 9
· leukemia ·
Expert Perspective on ASH 2014: Leukemia
Meir Wetzler, MD, FACP
More than 20,000 attendees from around the world gathered at the 2014 Annual Meeting and Exposition of the American Society of Hematology (ASH), which
convened on December 6, 2014, at the Moscone Center in San Francisco. The 4-day meeting is widely regarded as the foremost event in malignant and nonmalignant hematology for both phy-sicians and scientists working in the field.1
Abstracts presented at this year’s meeting signaled evolu-tions in treatment algorithms and provided attendees with a glimpse into what the future of clinical practice may look like, as heralded by the promise of exciting new agents in development. This review presents highlights of key abstracts in leukemia (see the January 2015 issue for highlights in lymphoma).
Sorafenib as Add-on to Standard Induction and Consolidation in AMLResults from the SORAMFL trial, which tested sorafenib versus placebo as add-on therapy to standard induction and consolida-tion treatment in patients 60 years or younger with acute myelog-enous leukemia (AML), indicated that the addition of sorafenib significantly prolonged event-free survival (EFS) and relapse-free survival (RFS) in this patient subset. There were no differences in overall survival (OS).2
Patients ranging in age from 18 to 60 years with newly diag-nosed AML were enrolled in the trial, which spanned 25 centers. All patients received 2 cycles of induction with daunorubicin (DA) plus cytarabine, followed by 3 cycles of high-dose cytarabine consolidation. Patients who showed no response following DA received a second induction with cytarabine plus mitoxantrone. All intermediate-risk and high-risk patients were scheduled to undergo allogeneic stem cell transplantation during first com-plete remission (CR).2
Patients were randomized to receive either sorafenib 800 mg/day (n = 134) or placebo (n = 133) as add-on to standard treat-ment in a double-blinded fashion. The trial’s primary endpoint was EFS, with an event being defined as failure to achieve a CR after induction, relapse, or death. Secondary endpoints included RFS, OS, CR rate, and incidence of adverse events (AEs).2
Rates of CR were similar between treatment arms; specifical-ly, 59% in the placebo arm versus 60% in the sorafenib arm (P = .764). After a median observation time of 36 months, medi-an EFS was 9.2 months in the placebo arm versus 20.5 months in the sorafenib arm, corresponding to a 3-year EFS of 22% ver-sus 40% (P = .013), respectively. Median RFS was 23 months after standard treatment plus placebo, but had not yet been reached after sorafenib treatment; this corresponded with 3-year RFS rates of 38% and 56% (P = .017), respectively. The 3-year OS rate was 56% with placebo versus 63% with sorafenib (P = .382); median OS had not yet been reached.2
Of note, in 46 FMS-like tyrosine kinase 3 (FLT3)-internal tan-dem duplication (ITD)-positive patients, no difference in EFS was observed; however, there was a trend toward a prolonged RFS and OS in favor of sorafenib. A possible explanation for the beneficial, nonspecific effects of sorafenib includes its effect on multiple kinases, such as vascular endothelial growth factor, platelet-derived growth factor, c-Kit, Raf kinases, and others.2
Intensification of DA in Induction in AMLData were also presented from the first randomized trial of 90 mg/m2 of DA versus 60 mg/m2 of DA in AML.3 Recent evidence has suggested improved rates of remission and OS from inten-sification of DA in induction with a higher dosage (90 mg/m2) versus the standard dosage (45 mg/m2) for patients with AML 17 to 65 years.4,5
The UK NCRI AML17 trial randomized 1206 patients (medi-an age 53 years; range, 16-72 years) with AML in a 1:1 fashion to 90 mg/m2 or 60 mg/m2 of DA on days 1, 3, and 5 in their first induction course, followed by 50 mg/m2 on days 1, 3, and 5 in their second course. No differences in remission rate were demonstrated, as remission was achieved in 81% of patients in the 90-mg/m2 cohort and 84% of patients in the 60-mg/m2
cohort (odds ratio [OR] 1.21, 0.90-1.64; P = .1). Two-year RFS was 52% versus 50% in the 90-mg and 60-mg arms, respective-ly (hazard ratio [HR] 1.06, 0.85-1.32; P = .6), and cumulative incidence of relapse was 37% in the 90-mg arm versus 41% in the 60-mg arm (HR = 1.01, 0.79-1.30; P = .9). Two-year OS was

10 www.ajho.com FEbRUARY 2015
· leukemia ·
59% versus 60% in the 90-mg and 60-mg arms, respectively (HR = 1.17, 0.95-1.44; P = .14), suggesting that the lower dose could be adopted without negatively affecting outcomes. Finally, subgroup analyses including age, karyotype, performance status, and FLT3-ITD/NPM1 genotypes did not show any benefit for the 90-mg dosage versus the 60-mg dosage.3
Azacitidine Versus Conventional Care RegimensA large phase 3 multicenter randomized trial, the AZA-AML-001 study, demonstrated that compared with conventional care regi-mens (CCR), treatment with azacitidine (AZA) prolonged medi-an OS by approximately 4 months (10.4 months vs 6.5 months; P = .1009) in older patients with newly diagnosed AML. Patients with AML with morphologic dysplastic changes (AML-MDC) comprised about 33% of participants in the trial.6
An international team of researchers sought to determine the effects of AZA versus CCR on OS, response, and safety in the subset of patients with AML-MDC in the AZA-AML-001 trial (n = 158), and to further analyze OS in patients with AML-MDC who had been preselected to receive low-dose cytarabine (LDAC) before randomization to AZA or CCR. The investigators found that the median OS in patients with AML-MDC was doubled with AZA versus CCR, 12.7 months versus 6.3 months, respec-tively (95% CI, 7.2-14.1; HR = 0.69, 0.48-0.98; P = .0357). One-year survival was also improved with AZA versus CCR, at 50.7% versus 33.8%, respectively (16.9% difference; 95% CI, 1.5-32.2). Rates of CR plus complete remissions with incomplete blood count recovery were 26.7% with AZA versus 19.3% with CCR. The investigators concluded that AZA was safe, effective, and well tolerated in this high-risk subset of patients with AML com-pared with CCR, which is frequently used in this setting.6 It will be interesting to see if AZA will replace LDAC in Europe follow-ing this trial.
Novel IDH2 InhibitorEarly-stage testing of a novel inhibitor of the IDH2 gene has shown promise in the treatment of leukemia and other hemato-logic blood cancers. IDH2 is an enzyme that converts isocitrate to a-ketoglutarate. IDH2 mutations cause decreased formation of a-ketoglutarate and increased formation of 2-hydroxyl glutarate, which acts as an oncometabolite by inducing epigenetic changes and impaired cell differentiation. AG-221 is a first-in-class, oral, potent, reversible, selective inhibitor of the IDH2 mutant en-zyme.7 At ASH 2014, data were presented from an ongoing phase 1, open-label, dose-escalation study of AG-221. Patients with ad-vanced IDH2 mutation-positive hematologic malignancies were administered AG-221 as a single agent once or twice daily in 28-day cycles. The study’s primary objectives were to determine the maximum tolerated dose (MTD) and safety, and to select a dos-age and schedule for expansion cohorts and future phase 2 trials. Secondary objectives included assessment of clinical activity by
investigators using the International Working Group Criteria, pharmacokinetics, and pharmacodynamics.8
Forty-eight patients have been enrolled since September 2013; 27 remain on treatment. To date, AG-221 has been well tolerat-ed, with MTD not yet reached, and the majority of reported AEs were grade 1 or 2. Nine patients have died, 8 within the first 28 days of receiving AG-221. One patient with severe pneumonia also died, with the death reported as possibly being related to the drug. Eleven serious AEs in 8 patients were reported as possibly drug related.8
Investigator-assessed objective responses have been observed in 20 patients. Responses have been durable, including complete remissions of up to 4.5 months. Although still early, these data suggest that mutant IDH2 is a valid therapeutic target.8
Fitness Criteria to Guide Treatment in Elderly With AMLThe use of intensive chemotherapy, nonintensive chemotherapy, or best supportive care to treat elderly patients with AML is a subject of ongoing debate. Although treatment choice is largely driven by a patient’s age, the role of fitness and comorbidities in treatment choice and outcome has garnered increasing attention in recent years.9 In 2013, Ferrara and colleagues proposed a set of objective criteria for defining patients as “fit” or “unfit” for intensive chemotherapy.10 In an effort to validate these criteria in the clinical setting, a team of Italian physicians utilized these criteria to perform a retrospective analysis of a population-based series of patients with AML.9
borlenghi and colleagues evaluated 350 patients 65 years or older who were diagnosed with AML at various hematologic cen-ters in Italy between January 2008 and May 2014; median age was 73 years. Using Ferrara’s criteria, the patients were classified as fit for intensive chemotherapy (fit), unfit for intensive chemo-therapy (unfit), or unfit for nonintensive chemotherapy (frail).9
Of the 350 evaluable patients, 170 (46.9%) were classified as fit, 140 (38.7%) were classified as unfit, and 40 (11%) were clas-sified as frail. Median OS of fit, unfit, and frail patients was 12.5 months, 3.7 months, and 1.8 months, respectively (fit vs others, P =. 0001; unfit vs frail, P =.049). Overall concordance between Ferrara’s fitness criteria and the treatment actually received by the patients was 80% (71% in fit, 88% in unfit, and 90% in frail patients).9
In this analysis, fitness level was significantly related to surviv-al. The median OS of patients receiving intensive chemotherapy, nonintensive chemotherapy, or best supportive care was 14.7 months, 14.2 months, and 4.2 months, respectively, in fit pa-tients (P <.0001), and 8.6 months, 8.9 months, and 2 months, respectively, in unfit patients (P <.0001). Median OS in frail pa-tients receiving nonintensive chemotherapy (n = 4) or best sup-portive care was 11.5 months and 2 months, respectively (not significant). The authors concluded that Ferrara’s fitness criteria appear to be useful for identifying patients likely to benefit from
VOL. 11, NO. 2 THE AMERICAN JOURNAL OF HEMATOLOGY/ONCOLOGY 11
ASH 2014
intensive or nonintensive chemotherapy as opposed to best sup-portive care, and for making decisions when treating elderly pa-tients with AML.9 A prospective study is needed to substantiate these findings.
Quizartinib in AMLFLT3-ITD mutations have been associated with early relapse and poor survival in AML. The novel agent quizartinib (formerly AC220) is a potent, targeted FTL3 inhibitor that selectively in-hibits FLT3 kinase activity. Gautem borthakur, MD, and col-leagues from MD Anderson Cancer Center in Houston present-ed data from a planned interim analysis of an ongoing phase 1/2 trial testing whether the addition of quizartinib to salvage thera-py with AZA or LDAC will improve response rates versus mono-therapy with either agent. The primary objective of the phase 1 trial was to determine the dose-limiting toxicity and MTD of the combination of quizartinib with either AZA or LDAC; the objective of the phase 2 trial was to determine the clinical activity of both combinations.11
At present, 26 patients have been enrolled, 18 to the AZA arm and 8 to the LDAC arm. Quizartinib 60 mg/day was selected as the recommended phase 2 dosage based on emerging results from a separate dose-finding study. Eighteen patients, all with FLT3-ITD mutations without D835 mutations, have responded, including 5 patients (63%) in the LDAC arm and 13 patients (72%) in the AZA arm. The overall response rate was 82% among patients with FLT3-ITD mutations (n = 22). These rates were higher than what was expected with either agent alone. Pa-tients continue to be enrolled to both arms of the trial.11
CAR-T CellsSome of the most exciting and interesting early-stage develop-ments concern the use of chimeric antigen receptor-modified T cells (CAR-T cells), which have demonstrated increasing po-tential for the treatment of various hematologic malignancies. Preclinical and clinical studies utilizing this type of adoptive im-munotherapy have achieved dramatic successes in the treatment
of AML, chronic lymphocytic leukemia (CLL), and solid tumor cancers, spurring ongoing investigations.12 by engineering T-cell function, as well as creating vigorous anti-tumor T-cell response and cancer-targeting memory T cells, it is hoped that this novel therapeutic approach may offer long-term disease control and possibly even curative potential.12
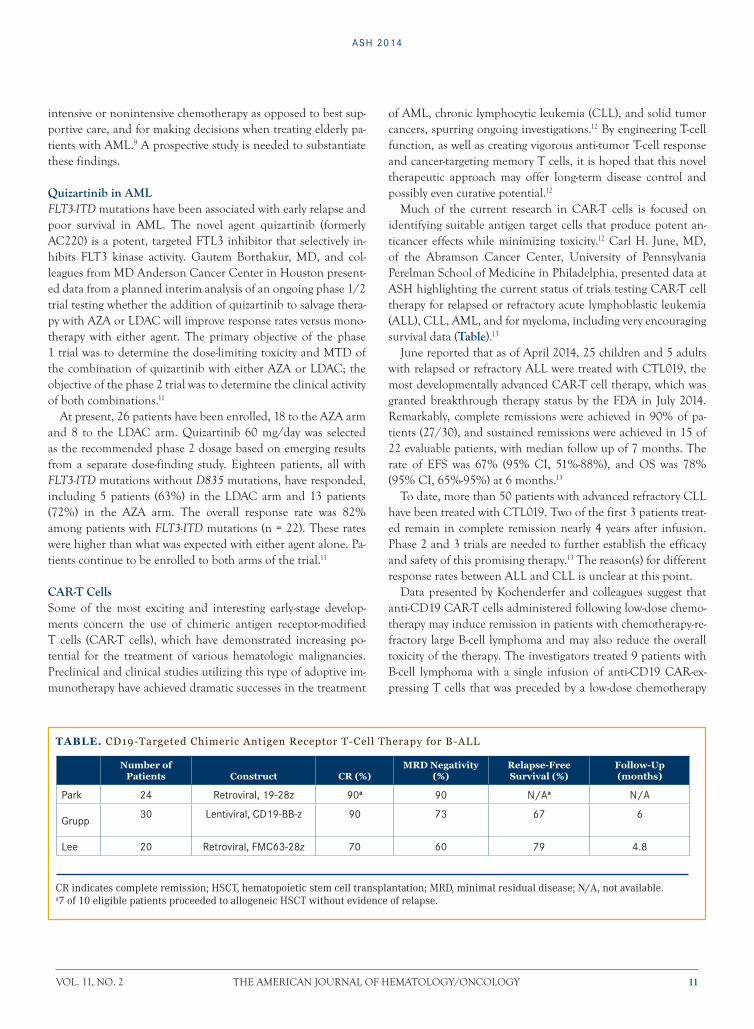
Much of the current research in CAR-T cells is focused on identifying suitable antigen target cells that produce potent an-ticancer effects while minimizing toxicity.12 Carl H. June, MD, of the Abramson Cancer Center, University of Pennsylvania Perelman School of Medicine in Philadelphia, presented data at ASH highlighting the current status of trials testing CAR-T cell therapy for relapsed or refractory acute lymphoblastic leukemia (ALL), CLL, AML, and for myeloma, including very encouraging survival data (Table).13
June reported that as of April 2014, 25 children and 5 adults with relapsed or refractory ALL were treated with CTL019, the most developmentally advanced CAR-T cell therapy, which was granted breakthrough therapy status by the FDA in July 2014. Remarkably, complete remissions were achieved in 90% of pa-tients (27/30), and sustained remissions were achieved in 15 of 22 evaluable patients, with median follow up of 7 months. The rate of EFS was 67% (95% CI, 51%-88%), and OS was 78% (95% CI, 65%-95%) at 6 months.13
To date, more than 50 patients with advanced refractory CLL have been treated with CTL019. Two of the first 3 patients treat-ed remain in complete remission nearly 4 years after infusion. Phase 2 and 3 trials are needed to further establish the efficacy and safety of this promising therapy.13 The reason(s) for different response rates between ALL and CLL is unclear at this point.
Data presented by Kochenderfer and colleagues suggest that anti-CD19 CAR-T cells administered following low-dose chemo-therapy may induce remission in patients with chemotherapy-re-fractory large b-cell lymphoma and may also reduce the overall toxicity of the therapy. The investigators treated 9 patients with b-cell lymphoma with a single infusion of anti-CD19 CAR-ex-pressing T cells that was preceded by a low-dose chemotherapy
Table. CD19-Targeted Chimeric Antigen Receptor T-Cell Therapy for B-ALL
Number of Patients Construct CR (%)
MRD Negativity (%)
Relapse-Free Survival (%)
Follow-Up (months)
Park 24 Retroviral, 19-28z 90a 90 N/Aa N/A
Grupp30 Lentiviral, CD19-BB-z 90 73 67 6
Lee 20 Retroviral, FMC63-28z 70 60 79 4.8
CR indicates complete remission; HSCT, hematopoietic stem cell transplantation; MRD, minimal residual disease; N/A, not available.a7 of 10 eligible patients proceeded to allogeneic HSCT without evidence of relapse.

12 www.ajho.com FEbRUARY 2015
· leukemia ·
regimen administered daily for 3 days (cyclophosphamide 300 mg/m2 and fludarabine 30 mg/m2). Eight of the 9 treated pa-tients had diffuse large b-cell lymphoma that was refractory to or that had relapsed less than 1 year after autologous stem cell transplantation—grim clinical scenarios with a median OS of less than 1 year.14
Despite their poor prognoses, 1 patient obtained a CR and 4 obtained partial responses, including resolution of large lym-phoma masses in some cases. Compared with previous studies that utilized high-dose chemotherapy prior to administration of anti-CD19 CAR-T cells, toxicity was reduced when CAR-T cells were infused after low-dose chemotherapy. There were no cases requiring vasopressor drugs or mechanical ventilation, and cyto-penias were mild.14
Blinatumomab in ALLThe detection of leukemic cells in bone marrow by polymerase chain reaction or flow cytometry in the presence of hematologic CR in ALL is known as minimal residual disease (MRD). Pa-tients with persistent or recurrent MRD after induction therapy are known to have a greater risk of relapse than those with no de-tectable MRD. When patients have MRD, the goal of treatment is to avoid hematologic relapse, reduce MRD load, and provide a bridge to subsequent hematopoietic stem cell transplantation (HSCT).15
blinatumomab is an investigational bi-specific T-cell (biTE) antibody construct that redirects CD3 T cells to CD19 target cells, resulting in serial lysis of CD19 b cells. In a phase 2 study of first-line blinatumomab in patients with MRD ALL (n = 21), 80% of evaluable patients achieved a complete MRD response.16 That trial was followed by bLAST, a confirmatory, single-arm, phase 2 study that evaluated the efficacy, safety, and tolerabili-ty of blinatumomab in patients with MRD ALL, the results of which were presented at ASH.15
bLAST enrolled patients 18 years or older with b-precursor ALL in hematologic CR (<5% blasts in bone marrow) after 3 or more intensive chemotherapy treatments and with MRD ≥10. blinatumomab 15 μg/m²/day was administered for 4 weeks by continuous IV infusion, followed by a 2-week treatment-free peri-od (1 cycle). Responders could receive up to 4 cycles of treatment or undergo HSCT after at least 1 cycle. Patients who experienced hematologic relapse discontinued treatment. The primary end-point was rate of complete MRD response; OS, RFS, duration of complete MRD response, and incidence and severity of AEs were secondary endpoints. OS and RFS will be analyzed after a minimum of 18 months of follow up.15
The trial enrolled 116 patients, each of whom received treat-ment. Median age was 45 years; 13% (15) patients were aged ≥65 years. As of February 2014, 74 patients had completed treatment (4 cycles or 1 cycle followed by HSCT) and 32 patients had dis-continued treatment due to AEs, disease relapse, or investigator
decision; an additional 79 patients were still alive and being fol-lowed. Three patients were excluded from the efficacy analysis: 1 patient had no central lab assay and 2 patients had assays with a sensitivity of 5 × 10–4.15
Among evaluable patients, 78% (88) had a complete MRD response following 1 cycle (95% CI, 69%-85%), confirming that the study met its primary objective. Two additional patients had a complete MRD response after more than 1 cycle of blinatu-momab. The complete MRD response rate across all cycles was 80%. All patients experienced at least 1 AE. The most common AEs, occurring in ≥20% of patients, included pyrexia (88%), headache (38%), tremor (29%), chills (25%), fatigue (24%), nausea (22%), and vomiting (22%). Sixty percent of patients ex-perienced serious AEs: 59% and 27% of patients had grade ≥3 and grade ≥4 AEs, respectively. Serious AEs occurring in ≥5% of patients were pyrexia (15%), tremor (7%), aphasia (5%), en-cephalopathy (5%), and overdose (5%). Two fatal AEs occurred on treatment, 1 of which (atypical pneumonia) was considered treatment-related. These results suggest that blinatumomab may have the potential to effectively eradicate MRD following inten-sive treatment.15 A large study of blinatumomab in relapsed/refractory CD19-positive ALL was recently published in Lancet Oncology17 and blinatumomab was recently approved by the FDA for relapsed/refractory ALL.
Adolescents and Young Adults (AYA) with ALLRetrospective analyses have shown that AYA with ALL have sig-nificantly improved survival when treated according to pediatric versus adult regimens. As such, the large, prospective C10403 US intergroup trial sought to evaluate the feasibility and effec-tiveness of treating AYA ALL patients (aged 16-39 years) using the Capizzi methotrexate arm of the successful Children’s On-cology Group regimen (COG AALL0232). EFS was the primary endpoint.18
AYA patients with newly diagnosed b-precursor ALL (b-ALL) or T-precursor ALL (T-ALL) were enrolled in the trial. The treat-ment protocol consisted of 5 intensive courses: remission induc-tion, remission consolidation, interim maintenance, delayed intensification, and prolonged maintenance therapy. Patients with M2 marrow response (>5% but <25% lymphoblasts) after remission induction received an extended remission induction course of therapy.18
From November 2007 through August 2012, 318 patients with a median age at diagnosis of 24 years were enrolled in the study; 22 patients withdrew prior to therapy. The majority of evaluable patients had b-ALL and were male (76% and 61%, respectively).18 Five deaths occurred that were deemed treatment-related: these included liver failure in 2 patients, both during induction; infec-tion in 1 patient during induction and 1 in consolidation; and ventricular arrhythmia in 1 patient during induction. Treatment toxicities were similar to those reported in the Capizzi metho-

VOL. 11, NO. 2 THE AMERICAN JOURNAL OF HEMATOLOGY/ONCOLOGY 13
ASH 2014
trexate arm of COG AALL0232, with an increased incidence of thrombosis and early hyperbilirubinemia.18
To date, 87 patients remain on treatment and 70 patients have died. The median EFS is 59.4 months (95% CI, 38.4 - not reached), and the 2-year EFS rate is 66% (95% CI, 60%-72%). Similar 2-year EFS rates were observed in b-ALL patients and T-ALL patients (65% and 68%, respectively). The 2-yr OS rate was 78% among b-ALL patients (95% CI, 72%-83%) and 80% for T-ALL (95% CI, 72%-84%).18
The investigators noted that the absence of detectable MRD was associated with 100% EFS (P = .0006). The improvements in clinical outcomes demonstrated in this trial are expected to form the basis for future trials, including those using novel agents to further improve survival for AYA with ALL.18 Another unique finding of this trial is the characterization of Philadelphia (Ph)-chromosome-like phenotype in 28% of the patients; their EFS was a mere 52% compared with 82% (P = .04) in patients without this phenotype. Novel approaches for Ph-like ALL are urgently needed.
Nilotinib + Chemotherapy for Ph+ ALLThe prognosis of elderly patients with Ph+ ALL has remained poor in spite of the high complete hematologic remission (CHR) rates achieved with imatinib-based treatment, largely due to the tendency to relapse in that patient subset. The potent AbL tyro-sine kinase inhibitor (TKI) nilotinib has been approved for the treatment of chronic and accelerated phase CML, but limited data on its efficacy in Ph+ ALL are available. To study the ac-tivity of an AbL-TKI regimen in the front-line setting, the Eu-ropean Working Group for Adult ALL developed a joint che-motherapeutic protocol for first-line therapy of elderly Ph+ ALL patients.19
Patients 55 years or older with Ph+ and/or BCR-ABL1-positive ALL were enrolled in the trial. The only prior treatments that were permitted were corticosteroids, single-dose vincristine, or 3 doses of cyclophosphamide. The trial’s primary endpoint was the rate of patients without an event at 12 months (an event was defined as relapse, death, serious AE, or treatment discon-tinuation). Secondary endpoints included EFS, OS, the rate of CHR after induction; death during induction or in CHR; and the rate of major molecular response or complete molecular re-sponse defined by BCRABL1/ABL1 ratios <0.1% and < 0.001%, respectively.19
As of August 2014, 47 patients with a median age of 66 years were enrolled. The CHR rate among patients evaluable for re-sponse (36) was 97%; 1 patient was refractory (3%). No patient died during induction therapy. After a median follow up of 211 days, 31 of 35 evaluable patients were in complete cytogenetic response and 4 patients had relapsed, 2 of whom had discon-tinued study treatment in order to undergo allogeneic stem cell transplant. Eight of 35 CR patients completed the consolidation
cycles and have entered maintenance phase; 5 patients have com-pleted protocol therapy. The rate of complete molecular remis-sion after induction was 30%, and 2 patients had undetectable BCR-ABL1 transcripts. During the consolidation phase, 42% of patients had a complete molecular remission, and BCR-ABL1 transcripts were undetectable in 29% of patients.
Tolerability was acceptable, with 34 serious AEs reported to date: 11 during induction, 16 during consolidation, 6 during the maintenance phase, and 1 following study discontinuation. Infectious events and neutropenic fever were the most common AEs. The investigators concluded that nilotinib combined with chemotherapy was well tolerated and highly effective, with a 97% CR in elderly patients with newly diagnosed Ph+ ALL. Molec-ular response rates were high, and MRD levels in responding patients have continued to decrease. This abstract suggests that nilotinib can become part of the armamentarium for Ph+ ALL.Affiliation: Meir Wetzler, MD, FACP, is chief, Leukemia Sec-tion, and professor of medicine in the Department of Medicine, at Roswell Park Cancer Institute, buffalo, NY.Disclosures: Dr Wetzler serves as a consultant to Novartis, Sig-ma-Tau, and Jazz Pharmaceuticals and is a principal investigator on studies with bristol-Myers Squibb and Teva. Writing assis-tance was provided by Kathleen Krafton, a freelance medical writer for AJHO. Ms Krafton has no relevant financial conflicts of interest to disclose.Acknowledgment of support: Supported partially by grants from the National Cancer Institute Grant CA16056, the Szefel Foundation, Roswell Park Cancer Institute, the Leonard S. Lu-Vullo Endowment for Leukemia Research, the Nancy C. Cully Endowment for Leukemia Research, the babcock Family Endow-ment, and the Heidi Leukemia Research Fund, buffalo, NY.Address correspondence to: Meir Wetzler, MD, FACP, Leu-kemia Section, Department of Medicine, Roswell Park Can-cer Institute, buffalo, NY 14263. Phone: 716-845-8447; email: [email protected].
REFERENCES1. American Society of Hematology. 56th ASH Annual Meet-ing to Highlight Cutting-Edge Research, Celebrate Major Mile-stones in Hematology. http://www.hematology.org/Newsroom/Press-Releases/2014/3448.aspx. Accessed January 12, 2014.2. Röllig C, Müller-Tidow C, Hüttmann A, et al. Sorafenib ver-sus placebo in addition to standard therapy in younger patients with newly diagnosed acute myeloid leukemia: results from 267 patients treated in the randomized placebo-controlled SAL-Soraml trial. Presented at: American Society of Hematology An-nual Meeting; December 6-9, 2014; San Francisco, CA.3. burnett AK, Russell N, Hills RK, et al. A randomised com-parison of daunorubicin 90mg/m2 vs 60mg/m2 in AML induc-tion: results from the UK NCRI AML17 trial in 1206 patients.

14 www.ajho.com FEbRUARY 2015
· leukemia ·
Presented at: American Society of Hematology Annual Meeting; December 6-9, 2014; San Francisco, CA.4. Fernandez HF, Sun Z, Yao X, et al. Anthracycline dose intensi-fication in acute myeloid leukemia. N Engl J Med. 2009;361:1249-1259.5. Löwenberg b, Ossenkoppele GJ, van Putten W, et al. High-dose daunorubicin in older patients with acute myeloid leuke-mia. N Engl J Med. 2009;361:1235-1248.6. Seymour JF, Döhner H, Aleksandra butrym A, et al. Azacit-idine (AZA) versus conventional care regimens (CCR) in older patients with newly diagnosed acute myeloid leukemia (>30% bone marrow blasts) with myelodysplasia-related changes: a sub-group analysis of the AZA-AML-001 trial. Presented at: Amer-ican Society of Hematology Annual Meeting; December 6-9, 2014; San Francisco, CA.7. Grisham J. Can cells be turned from cancerous to normal? http://www.mskcc.org/blog/can-cells-be-turned-cancerous-nor-mal. Accessed January 12, 2015.8. Stein E, Altman JK, Collins R, et al. AG-221, an oral, selective, first-in-class, potent inhibitor of the IDH2 mutant metabolic en-zyme, induces durable remissions in a phase I study in patients with IDH2 mutation positive advanced hematologic malignan-cies. Presented at: American Society of Hematology Annual Meeting; December 6-9, 2014; San Francisco, CA.9. borlenghi E, Pagani C, basilisc C, et al. Validating the pa-tient’s “fitness” criteria proposed to guide treatment decision in elderly AML: a multicenter study on a population-based series of 362 patients by the network “Rete Ematologica Lombarda” (REL). Presented at: American Society of Hematology Annual Meeting; December 6-9, 2014; San Francisco, CA.10. Ferrara F, barosi G, Venditti A, et al. Consensus-based defi-nition of unfitness to intensive and nonintensive chemotherapy in acute myeloid leukemia: a project of SIE, SIES and GITMO group on a new tool for therapy decision making. Leukemia. 2013;27:997-999.11. borthakur G, Kantarjian HM, O’brien S, et al. The combi-nation of quizartinib with azacitidine or low dose cytarabine is highly active in patients (pts) with FLT3-ITD mutated myeloid leukemias: interim report of a phase I/II trial. Presented at: American Society of Hematology Annual Meeting; December 6-9, 2014; San Francisco, CA.12. American Society of Hematology. Annual meeting: special scientific symposia. http://www.hematology.org/Annual-Meet-ing/Program/Special-Scientific-Symposia.aspx. Accessed January 10, 2014.13. June C. Therapeutic efficacy of chimeric antigen receptor T cells. Presented at: American Society of Hematology Annual Meeting; December 6-9, 2014; San Francisco, CA.14. Kochenderfer JN, Somerville R, Lu L, et al. Anti-CD19 CAR T cells administered after low-dose chemotherapy can induce remissions of chemotherapy-refractory diffuse large b-cell lym-
phoma. Presented at: American Society of Hematology Annual Meeting; December 6-9, 2014; San Francisco, CA.15. Goekbuget N, Dombret H, bonifacio M, et al. bLAST: a confirmatory, single-arm, phase 2 study of blinatumomab, a bispecific T-cell engager (biTE ) antibody construct, in patients with minimal residual disease b-precursor acute lymphoblastic leukemia (ALL). Presented at: American Society of Hematology Annual Meeting; December 6-9, 2014; San Francisco, CA.16. Topp MS, Gökbuget N, ZugmaierG, et al. Long-term fol-low-up of hematologic relapse-free survival in a phase 2 study of blinatumomab in patients with MRD in b-lineage ALL. Blood. 2012;120(26):5185-5187.17. Topp MS, Gökbuget N, Stein AS, et al. Safety and activity of blinatumomab for adult patietns with relapsed or refractory b-precursor acute lymphoblastic leukaemia: a multicentre, sin-gle-arm, phase 2 study. Lancet Oncol. 2015;16:57-66.18. Stock W, Luger SM, Adrani PS, et al. Favorable outcomes for older adolescents and young adults (AYA) with acute lym-phoblastic leukemia (ALL): early results of U.S. intergroup trial C10403. Presented at: American Society of Hematology Annual Meeting; December 6-9, 2014; San Francisco, CA.19. Ottmann OG, Pfeifer H, Cayuela J-M, et al. Nilotinib (Ta-signa®) and chemotherapy for first-line treatment in elderly patients with De Novo Philadelphia chromosome/bCR-AbL1 positive acute lymphoblastic leukemia (ALL): a trial of the Euro-pean working group for adult ALL (EWALL-PH-02). Presented at: American Society of Hematology Annual Meeting; December 6-9, 2014; San Francisco, CA.

VOL. 11, NO. 2 THE AMERICAN JOURNAL OF HEMATOLOGY/ONCOLOGY 15
· prostate cancer ·
Anti-Androgen Therapies for Prostate Cancer: A Focused Review
Nischala Ammannagari, MD, and Saby George, MD, FACP
IntroductionAmong men in the United States, prostate cancer is the most common malignancy and the second leading cause of mortality behind lung cancer.1 Since the discovery of the androgen depen-dence of prostate cancer in 1941 by Huggins and colleagues,2 androgen deprivation therapy (ADT) has remained the main-stay of treatment for prostate cancer.3 A better understanding of the androgen receptor (AR) signaling pathway and mechanisms of resistance to castration over the past decade has led to the discovery of novel AR targeting agents such as abiraterone and enzalutamide. This article is a focused review on novel targeted therapies in castration-resistant prostate cancer (CRPC) and bio-markers of resistance to such therapies.
Androgen Receptor Signaling and CRPC In a normal prostate, androgen receptors play a major role in the development of the prostate and regulating of the genes en-coding protein transcription for normal prostate function. In prostate cancer, increased levels of androgens in the tumor cells promote AR signaling.4 This in turn modulates gene expression associated with cell-cycle regulation, growth, and survival, and promotes the expression of oncogenic fusion genes. Tradition-
ally, treatments have attempted to decrease the amount of cir-culating androgens and AR signaling through the activation of the AR. This was achieved by surgical and chemical castration.3,4
However, patients frequently develop resistance to castration, and several different mechanisms have been attributed to the development of such resistance. Studies over the past few years have shown that in CRPC the levels of intratumoral and adre-nal androgens, namely testosterone and dihydrotestosterone, are similar to those in the noncastrated prostate, and therefore, there is still a role for secondary hormonal manipulation.4-6 In addition to these residual androgens, we also know that overex-pression or gene amplification of AR, point mutations in the AR ligand-binding domain (LBD), and expression of active AR splic-ing variants lead to maintained AR signaling in CRPC, causing first-generation anti-androgens to become partial AR agonists.4,6
The two main classes of anti-androgen therapies include an-drogen biosynthesis inhibitors and androgen receptor blockers (Figure 1). The drugs reviewed in this article in each of these classes are abiraterone and enzalutamide.
Androgen Biosynthesis Inhibitors A microsomal enzyme called CYP17 is essential for the biosyn-thesis of androgens and adrenal hormones. It acts by catalyzing 2 crucial steroid reactions: (1) hydroxylation of pregnenolone and progesterone at the C17 position to generate 17α-hydroxy-pregnenolone and 17α-hydroxyprogesterone, respectively; and (2) cleavage of the C17–C20 bond of 17α-hydroxypregnenolone and 17α- hydroxyprogesterone to form dehydroepiandroste-
Abstract
Among men in the United States, prostate cancer is the
most common malignancy and the second leading cause
of mortality next to lung cancer. Androgen deprivation
is the mainstay of treatment, and an increased under-
standing of the androgen receptor signaling pathway
and mechanisms of resistance to castration over the past
decade has led to the discovery of novel agents such as
abiraterone and enzalutamide, which target androgen re-
ceptor signaling. This article is a focused review on novel
targeted therapies in castration-resistant prostate cancer
and biomarkers of resistance to such therapies.
Key words: anti-androgen therapy, enzalutamide,
abiraterone, AR-V7, castration-resistant prostate cancer
figure 1. Different Classes of Anti-Androgen Therapy
Androgen biosynthesis
• Ketoconazole• Abiraterone• TAK-700• TOK-001
• Bicalutamide• Nilutamide• Flutamide• Enzalutamide• ARN-509
Androgen receptor blockers

16 www.ajho.com FEBRUARY 2015
· prostate cancer ·
rone (DHEA) and androstenedione, respectively.4,6 Inhibition of CYP17 thus causes decreased androgen synthesis, offering a therapeutic target in prostate cancer. Antifungal and nonspecific CYP17 inhibitors such as ketoconazole and novel, highly specific CYP17 inhibitors such as abiraterone, TAK-700, and TOK-001 fall under this category.4-6
Abiraterone: This is a highly selective CYP17 inhibitor that caus-es a decrease in adrenal androgens, thus indirectly inhibiting the AR signaling pathway. Two large clinical trials, COU-AA-301 and COU-AA-302, resulted in approval of abiraterone (Zytiga) in the management of CRPC in the postdocetaxel and chemo-therapy-naïve setting, respectively.7,8
COU-AA-301 was a randomized phase 3 trial that enrolled a total of 1195 patients with metastatic CRPC who were pre-viously treated with docetaxel.7 Patients were randomized in a 2:1 ratio to abiraterone acetate 1000 mg with prednisone 5 mg twice daily and placebo with prednisone arms, respectively, with a primary endpoint of overall survival (OS) and secondary endpoints of time to prostate-specific antigen (PSA) progression, progression-free survival (PFS), and PSA response rate. The study demonstrated prolonged OS in the abiraterone acetate-plus-pred-nisone group compared with the placebo-plus-prednisone group (14.8 months vs 10.9 months; hazard ratio [HR] = 0.65; 95% confidence interval [CI], 0.54-0.77; P <.001). The secondary end-points of time to PSA progression (10.2 vs 6.6 months; P <.001), PFS (5.6 months vs 3.6 months; P <.001), and PSA response rate (29% vs 6%; P <.001) favored the treatment group as well. The drug was reasonably well tolerated, with common adverse events (AEs) including fatigue, anemia, nausea, pain, arthralgia, edema, and constipation. This study led to approval of abiraterone in
treatment in the postdocetaxel setting of CRPC. The COU-AA-302 trial was a phase 3 randomized, dou-
ble-blind, placebo-controlled, multicenter study that compared the clinical benefit of abiraterone acetate plus prednisone with placebo plus prednisone in asymptomatic or mildly symptom-atic patients with metastatic CRPC prior to treatment with docetaxel.8 A total of 1088 patients were randomized in a 1:1 ra-tio to abiraterone-prednisone and placebo-prednisone groups, re-spectively. The co-primary endpoints were radiographic PFS and OS; key secondary outcome measures included time to opiate use for cancer-related pain, time to initiation of chemotherapy, time to deterioration of ECOG performance status by ≥1, and time to PSA progression. Although interim analysis at a median follow-up of 29 months showed no statistically significant OS benefit, results of the final analysis presented at the European Society for Medical Oncology (ESMO) 2014 Congress showed that at a median follow-up of 49.2 months, patients in the abi-raterone arm achieved a median OS of 34.7 months compared with 30.3 months in the placebo arm (HR = 0.81; 95% CI, 0.70-0.93; P = .0033). In addition, a significant improvement in all secondary endpoints, including median time to opiate use for cancer-related pain, time to initiation of cytotoxic chemother-apy, and time to PSA progression, was also noted in the abi-raterone-placebo group. The AE profile was very similar to that of COU-AA-301 study.
Androgen Receptor BlockersThe limitations of first-generation anti-androgens such as bi-calutamide, nilutamide, and flutamide, as just described, formed the basis for the preclinical development of second-generation anti-androgens such as enzalutamide and ARN-509.
Enzalutamide: Enzalutamide is a direct AR blocker that binds with a high affinity to the ligand-binding domain of the AR (Fig-ure 2). It prevents nuclear translocation of the AR, DNA bind-ing, and co-activator recruitment of the ligand-receptor complex. Two clinical trials, AFFIRM9 and PREVAIL,10 were noteworthy in approval of enzalutamide as a treatment option in metastatic CRPC.
The AFFIRM trial was a phase 3 randomized, double-blind, placebo-controlled trial that evaluated whether enzalutamide prolonged survival in patients with CRPC in the postchemother-apy setting.9 A total of 1199 men with CRPC after chemother-apy were randomly assigned in a 2:1 ratio to receive either oral enzalutamide 160 mg per day or placebo. The primary endpoint was OS, and the key secondary endpoints included reduction in PSA level by 50% or more, soft-tissue response rate, quality-of-life response rate, time to PSA progression, time to skeletal-related events (SREs), and radiographic PFS. The study demonstrated significantly prolonged survival with enzalutamide in the CRPC setting after docetaxel when compared with placebo (18.4 months
figure 2. Mechanism of Action of Enzalutamide
Inhibitsandrogens
binding to ARCellCytoplasm
CellNucleus
InhibitsAR nuclear
translocation
InhibitsAR mediatedDNA binding

VOL. 11, NO. 2 THE AMERICAN JOURNAL OF HEMATOLOGY/ONCOLOGY 17
Anti-Androgen therApies for prostAte CAnCer
vs 13.6 months; HR for death in the enzalutamide group, 0.63; 95% CI, 0.53-0.75; P <.001). In addition, it demonstrated the superiority of enzalutamide over placebo with respect to all sec-ondary endpoints. AEs of fatigue, diarrhea, and hot flashes were higher in the enzalutamide group, and seizures were reported in 5 patients (0.6%) receiving enzalutamide.
The PREVAIL study, a phase 3 double-blind study, assigned a total of 1717 patients with chemotherapy-naïve CRPC after failing standard ADT to receive either enzalutamide or placebo once daily in a 1:1 ratio.10 The co-primary endpoints were radio-graphic PFS and OS; some of the key secondary endpoints in-cluded the time to initiation of cytotoxic chemotherapy, time to first SRE, soft-tissue response, time to PSA progression, and PSA response. The study was stopped after a planned interim analysis conducted when 540 deaths had been reported. The enzalut-amide group demonstrated a significantly delayed radiographic disease progression (81% risk reduction; HR = 0.19; 95% CI, 0.15-0.23; P <.001) and improved OS (72 % vs 63%, with a 29% reduction in the risk of death; HR = 0.71; 95% CI, 0.60-0.84; P <.001). In addition, superiority of enzalutamide over placebo was shown with respect to all secondary endpoints. The drug was generally well tolerated, with common side effects being fatigue and hypertension. This study demonstrated that enzalutamide added to ADT at progression provided meaningful clinical ben-efit to men with chemotherapy-naïve metastatic prostate cancer.
Biomarkers of Resistance FormationAlthough secondary hormonal manipulation by these novel an-ti-androgen agents works in CRPC, patients tend to develop re-sistance. Over the years, several mechanisms of development of resistance have been proposed and tested in preclinical models as well as patient samples.
Sawyers and his group raised the possibility that in preclinical prostate cancer models, enzalutamide resistance may be medi-ated by glucocorticoid receptors (GRs).11 In animal models, the investigators were able to show that the GR is upregulated in prostate cancer cell lines and that dexamethasone reverses en-zalutamide-induced growth inhibition. They also reported a cor-relation between GR expression in patient-derived prostate can-cer specimens and clinical response to enzalutamide. However, the current clinical evidence for GR mediating drug resistance in patients with CRPC is very limited at best.
Sharifi and his group have proposed that resistance to abi-raterone may develop because of continued intratumoral ste-roidogenesis by a backdoor pathway despite strong CYP17A1 inhibition.12 They were able to show that in addition to CY-P17A1 inhibition, abiraterone can competitively inhibit 3β-hy-droxysteroid dehydrogenase (3β-HSD), which has been reported to convert DHEA to ASD, which is then 5α-reduced by 5α-re-ductase 1 to 5α-dione, and then DHT in cell lines and LAPC4 xenografts in rodent models. As a result, it was proposed that
the second, weaker action of blocking 3β-HSD enzymatic activity may perhaps be overcome by increasing abiraterone drug expo-sure by administration with a high-fat meal, increasing dosage of the drug, or both.
AR-V7: Androgen receptor variant 7 (AR-V7) is an AR iso-form encoded by splice variant 7. This spliced variant encodes a truncated AR protein that lacks the C-terminal LBD, but re-mains constitutively active as a transcription factor and is capable of promoting activation of target genes (Figure 3).13,14 The ex-pression of AR-V7 is increased by nearly 20-fold in CRPC tumor cells in certain patients. Because enzalutamide primarily inter-acts with the LBD of AR, it is expected that AR-V7 without LBD causes enzalutamide resistance. Furthermore, since abiraterone causes ligand depletion, it is expected that abiraterone will not work in the presence of ligand-independent AR-V7 protein.13,14
Prospective Study of AR-V7 Status in CRPC A prospective study was recently conducted to evaluate whether detection of AR-V7 in circulating tumor cells (CTCs) in patients with CRPC was associated with resistance to enzalutamide or abiraterone.14 A total of 31 enzalutamide-treated patients and 31 abiraterone-treated patients with metastatic CRPC were enrolled prospectively into this study, and a quantitative reverse-transcrip-tion polymerase chain reaction (qRT-PCR) showed that 39% and
figure 3. Full Length AR and Truncated AR (AR-V7)
(Courtesy of Emmanuel Antonarakis)
Full-Length AR (AR-FL)
AR-V7: Truncated, Lacks LBD
NTD DBD Hinge LBD
Activation Function 1:Required for transcriptional
activity
DBDNTD Hinge LBD
Androgen (DHT)Antiandrogens (AA)
CRPC contain variants which lack the LBD
No current therapies can inhibit because they work
through the LBD
AA
DHTCBP
CBP

18 www.ajho.com FEBRUARY 2015
· prostate cancer ·
19%, respectively, had detectable AR-V7. Associations between AR-V7 status and PSA response rates (the primary endpoint), PSA PFS, clinical or radiographic PFS, and OS (secondary end-points) were evaluated. CTC samples were collected at 3 time points: pretreatment baseline status; at time of response to en-zalutamide or abiraterone; and at time of resistance to enzalut-amide or abiraterone.
In the enzalutamide-treated patients, AR-V7-positive patients had lower PSA response rates than AR-V7-negative patients (0% vs 53%; P = .004); and shorter PSA PFS (median, 1.4 months vs 6.0 months; P <.001), clinical or radiographic PFS (median, 2.1 months vs 6.1 months; P <.001), and OS (median, 5.5 months vs not reached; P =.002).
Similarly, in the abiraterone-treated patients, AR-V7-positive patients had lower PSA response rates than AR-V7-negative pa-tients (0% vs 68%; P = .004); and shorter PSA PFS (median, 1.3 months vs not reached; P <.001), clinical or radiographic PFS (median, 2.3 months vs not reached; P <.001), and OS (median, 10.6 months vs not reached; P = .006). Interestingly, out of 42 patients who were AR-V-negative at baseline, 6 patients (14%) “converted” to AR-V7-positive on subsequent samples, and the clinical outcomes of men who “converted” from AR-V7-negative to -positive were intermediate.
The pearls from this study are that the detection of AR-V7 may be associated with primary and acquired resistance to enzalut-amide and abiraterone; patients with CRPC who have detect-able AR-V7 in CTC samples can be steered away from receiving AR-targeting drugs and offered alternative treatments instead.
Ongoing Clinical TrialsSeveral ongoing clinical trials are looking at the combination and sequencing of enzalutamide and abiraterone in both CRPC and hormone-naïve prostate cancer, some of which are listed in the Table.
Most recently, in a press release, the results of the phase 2 TER-RAIN trial15 performing a head-to-head comparison between en-zalutamide and bicalutamide in metastatic prostate cancer were announced (NCT01288911). This trial enrolled 375 patients in North America and Europe who had metastatic prostate can-cer that had progressed despite luteinizing hormone-releasing hormone (LHRH) analogue therapy or surgical castration, and demonstrated a statistically significant increase in PFS in the en-zalutamide group compared with bicalutamide (15.7 months vs 5.8 months; HR = 0.44; 95% CI, 0.34-0.57; P <.0001).
Conclusion Targeting AR remains a viable strategy even after development of castration resistance.4 Androgen biosynthesis inhibition through abiraterone and direct AR blockade with second-generation an-ti-androgens such as enzalutamide have demonstrated a survival advantage in the CRPC setting, in both postdocetaxel as well as chemotherapy-naïve patients.7-10 Use of biomarkers of therapeu-tic resistance such as AR-V7 will help us identify which patients will not benefit from these targeted agents (ie, enzalutamide and abiraterone), thereby allowing us to offer alternative treatments to these patients.13,14 Given the lack of C-terminal LBD in this common AR variant, AR-V7 detection could also lead to devel-opment of novel N-terminal LBD targeted AR-blocking agents.
Interestingly, AR expression has been shown to be important in preventing the differentiation of epithelial prostate cancer cells into a neuroendocrine phenotype associated with tumor progres-sion. The unopposed continuous use of secondary hormonal manipulation by various anti-androgen therapies has been felt to be responsible for the emergence of this aggressive phenotype.16,17 For these reasons, it needs to be established whether the earlier use of secondary hormonal manipulation in the CRPC setting indeed could lead to poorer outcomes.
Table. Ongoing Clinical Trials of Enzalutamide and Abiraterone Sequences and Combinations
NCT Identifier Trial Description
NCT02125357 Randomized Phase 2 Study of Sequencing Abiraterone and Enzalutamide in mCRPC
NCT01650194 Phase 2 Study Determining Safety and Tolerability of Enzalutamide in Combination With Abiraterone in Bone Metastatic CRPC
NCT01949337 Phase 3 Trial of Enzalutamide Versus Enzalutamide plus Abiraterone and Prednisone for mCRPC
NCT02268175 Phase 2 Randomized Study of Enzalutamide+Leuprolide Versus Enzalutamide+Leuprolide+Abiraterone Acetate+Prednisone as Neoadjuvant Therapy for High-Risk Prostate Cancer Undergoing Prostatectomy
NCT00268476 A multistage, multiarm randomized trial comparing androgen suppression-based therapy alone versus combined with Zoledronic Acid, Docetaxel, Prednisolone, Celecoxib, Abiraterone, Enzalutamide and/or Radiotherapy in patients with locally advanced or metastatic prostate cancer (STAMPEDE)
Source: ClinicalTrials.gov

VOL. 11, NO. 2 THE AMERICAN JOURNAL OF HEMATOLOGY/ONCOLOGY 19
Anti-Androgen therApies for prostAte CAnCer
Affiliations: Nischala Ammannagari, MD, and Saby George, MD, FACP, are from Roswell Park Cancer Institute, Buffalo, NY.Disclosures: Dr Ammannagari reports no relevant conflicts of interest to disclose. Dr George has served as a consultant or on a paid advisory board for Novartis, Bayer, Astellas, and Sanofi.Address correspondence to: Nischala Ammannagari, MD, Ros-well Park Cancer Institute, Elm and Carlton St, Buffalo, NY 14263. Email: [email protected].
REFEREnCES1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11-30.2. Huggins C., Stephens, RE, Hodges CV. Studies on prostate cancer II. The effects of castration on advanced carcinoma of the prostate. Arch Surg. 1941;43:209-223. 3. Lytton, B. Prostate cancer: a brief history and the discovery of hormonal ablation treatment. J Urol. 2001;165:1859-1862.4. Wong YN, Ferraldeschi R, Attard G, de Bono J. Evolution of androgen receptor targeted therapy for advanced prostate cancer. Nat Rev Clin Oncol. 2014;11(6):365-376.5. Attard G, Richards J, de Bono JS. New strategies in metastatic prostate cancer: targeting the androgen receptor signaling path-way. Clin Cancer Res. 2011;17(7):1649-1657.6. Ahmed A, Ali S, Sarkar FH. Advances in androgen receptor tar-geted therapy for prostate cancer. J Cell Physiol. 2014;229(3):271-276.7. de Bono JS, Logothetis CJ, Molina A, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364(21):1995-2005.8. Ryan CJ, Smith MR, de Bono JS, et al. Abiraterone in meta-static prostate cancer without previous chemotherapy. N Engl J Med. 2013;368(2):138-148.9. Scher HI, Fizazi K, Saad F, et al. Increased survival with en-zalutamide in prostate cancer after chemotherapy. N Engl J Med. 2012;367(13):1187-1197. 10. Beer TM, Armstrong AJ, Rathkopf DE, et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med. 2014;371(5):424-433.11. Arora VK, Schenkein E, Murali R, et al. Glucocorticoid re-ceptor confers resistance to antiandrogens by bypassing andro-gen receptor blockade. Cell. 2013;155(6):1309-1322. 12. Li R, Evaul K, Sharma KK, et al. Abiraterone inhibits 3β-hy-droxysteroid dehydrogenase: a rationale for increasing drug ex-posure in castration-resistant prostate cancer. Clin Cancer Res. 2012;18(13):3571-3579.13. Haile S, Sadar MD. Androgen receptor and its splice variants in prostate cancer. Cell Mol Life Sci. 2011;68(24):3971-3981.14. Antonarakis ES, Lu C, Wang H, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014;371(11):1028-1038.
15. Baskin-Bey E, Shore N, Barber K, et al. TERRAIN: a ran-domized, double-blind, phase II study comparing MDV3100 with bicalutamide (Bic) in men with metastatic castrate-resis-tant prostate cancer (CRPC). J Clin Oncol. 2012;30(suppl; abstr TPS4698).16. Frigo DE, McDonnell DP. Differential effects of prostate cancer therapeutics on neuroendocrine transdifferentiation. Mol Cancer Ther. 2008;7(3):659-669.17. Lipianskaya J, Cohen A, Chen CJ, et al. Androgen-depriva-tion therapy-induced aggressive prostate cancer with neuroendo-crine differentiation. Asian J Androl. 2014;16(4):541-544.

20 www.ajho.com february 2015
· breast cancer ·
efficacy of Very-Low-Dose Capecitabine in Metastatic breast Cancer
Caitlin bertelsen, MD, Lingyun Ji, MS, agustin a. Garcia, MD, Christy russell, MD, Darcy Spicer, MD, richard Sposto, PhD, and Debu Tripathy, MD
Introductionbreast cancer causes approximately 40,000 deaths annually in the united States.1 The majority of these deaths occur in women with metastatic breast cancer (MbC), reflecting the incurability of MbC and the need for improved treatment. Currently, treat-ment for MbC is palliative and involves balancing improvement in cancer symptoms and delay of progression against side effects of therapy.2
Capecitabine is an orally administered prodrug of 5-fluoroura-cil (5-fu) that is activated to 5-fu preferentially in tumor tissue due to increased expression of thymidine phosphorylase in tu-mor tissue, which contributes to the drug’s specificity and action against tumor cell proliferation.3 Capecitabine was first approved for MbC in 1998 in patients pretreated with anthracyclines and taxanes. It was later approved in combination with docetaxel af-ter the combination resulted in improved overall survival (OS) when compared with docetaxel alone.4,5 Since then, capecitabine has come into wide use in MbC and is now approved both as monotherapy and in combination with docetaxel.6
Capecitabine was approved for use at a starting dosage of 1250 mg/m2 twice daily, with a dosing schedule of 14 days on followed by 7 days off despite frequent treatment-limiting toxicities, pri-marily hand-and-foot syndrome, stomatitis, and diarrhea at this dosage. These side effects are typically managed by dosage reduc-tion or starting at a lower dosage despite a lack of evidence from phase 3 randomized clinical trials to validate the efficacy of these lower dosages.
Since the approval of capecitabine, moderate dosage reduc-tions have been studied, either retrospectively7,8 or in small pro-spective trials9-11 in order to assess both the efficacy and frequen-cy of adverse effects (aes) associated with lower dosages. Other studies have examined the efficacy of dosages of capecitabine as low as 825 mg/m2 twice daily,12 suggesting that this dosage is no less efficacious than the full dosage. altering the treatment schedule has also been investigated as a method of reducing drug-associated toxicity. The administration of capecitabine via “continuous” dosing (without a 7-day rest period) using dosages as low as 650 mg/m2 twice daily was recently shown to be supe-rior in both tolerability and efficacy to classical cyclophospha-
Abstract Background: The FDA-approved dosage of capecitabine,
1250 mg/m2 twice daily, is often associated with treat-
ment-limiting toxicities. Clinical experience and published
reports suggest that lower starting dosages of capecit-
abine can be as effective as the approved dosage. In this
retrospective analysis we compared the efficacy of sig-
nificantly lower dosages of capecitabine with the FDA-ap-
proved dosage, using previously published results as
comparators.
Patients and Methods: We performed a retrospective co-
hort analysis of patients treated at University of Southern
California hospitals who received capecitabine as the first,
second, or third line of chemotherapy for metastatic or
unresectable locally advanced breast cancer to determine
the progression-free survival (PFS) associated with low
starting dosages.
Results: Patients (n = 84) received a median capecitabine
dosage of 565 mg/m2 twice daily, mostly administered as
a flat dosage (not adjusted for body surface area) of 1000
mg twice daily. The median PFS among patients with mea-
surable disease (n = 62; 74% of patients) was 4.1 months
(95% confidence interval, 2.9-5.7), which was similar to
the median PFS values (4.4 months; 4.2 months) for sin-
gle-agent capecitabine reported in the 2 major trials with
similar eligibility criteria. Furthermore, only 2 patients
(2.4%) discontinued capecitabine due to toxicity, support-
ing our hypothesis that starting treatment at low dosages
minimizes side effects while preserving efficacy.
Conclusions: Our results provide evidence that very low
dosages of capecitabine are efficacious in treating meta-
static breast cancer. Large-scale randomized, controlled
trials testing lower starting dosages of capecitabine are
necessary in order to firmly establish an optimally effec-
tive and well-tolerated dosage.
Key words: Capecitabine, chemotherapy, metastatic
breast cancer, drug toxicity, dose intensity

VOL. 11, NO. 2 THe aMerICaN JOurNaL Of HeMaTOLOGy/ONCOLOGy 21
Very-Low-Dose CapeCitabine in MbC
mide, methotrexate, and fluorouracil as first-line chemotherapy for MbC.13 another series of studies demonstrated tolerability of a 7-days-on, 7-days-off “dose-dense” regimen of capecitabine,14 as well as efficacy of this alternate schedule in combination with lapatinib15 or bevacizumab16 that was comparable to results of prior trials using the traditional 14-days-on, 7-days-off schedule.
The question of dosing is further complicated by the fact that fluoropyrimidine pharmacokinetics vary significantly between patients17 and demographic groups,18 such that precise dosing based on body surface area (bSa) may be unnecessary. The con-cept that variations in thymidine phosphorylase polymorphisms may explain differences in patients’ responses to the drug in both tumor and extratumoral tissues was proposed by Kaufmann et al9 as a rationale for the observed correlation between the devel-opment of hand-foot syndrome with time to progression (TTP) on capecitabine therapy. These findings provide hope that in the future, more objective measures will be used to individualize treatment and dosing regimens. Currently, however, selection of patients for treatment with capecitabine and modifications in dosing rely heavily on clinical judgment. The lack of consensus on an appropriate starting dosage of capecitabine has resulted in the use of myriad different dosing regimens and schedules in recent phase 1 and 2 clinical trials, many of which have been shown to be comparable in efficacy to standard dosing. addi-tionally, several retrospective studies have determined that lower dosages of capecitabine possess efficacy comparable to that of the full dosage, with less significant toxicity.7,8
at the university of Southern California (uSC) hospitals, defined as Los angeles County (LaC) + uSC Medical Center and uSC Norris Comprehensive Cancer Center, capecitabine is routinely prescribed at dosages as low as 600 mg/m2 twice daily, with a majority of patients receiving a flat dosage (not adjusted for bSa) of 1000 mg twice daily, lower than previously published series. In addition to the existing evidence of efficacy of lower dosages of capecitabine, the rationale for this practice is that the use of lower dosages facilitates patient adherence to treatment, as providers at the LaC+uSC Medical Center often encounter challenges with compliance and observation of follow-up ap-pointments among the underserved patient population treated at this hospital.
We sought to compare outcomes of patients at the uSC hospi-tals who were treated with very low dosages with published trials using standard fDa-approved dosing. a secondary aim was to identify clinical predictors of PfS, including dosage of capecit-abine, within our cohort. In order to best approximate the clin-ical population treated with capecitabine, we chose comparator studies that included patients with MbC treated with this agent in up to 3 lines of therapy.19,20
Patients and MethodsPatientsunder an institutional review board-approved protocol, we used electronic medical records (eMrs), pharmacy records, and care providers’ patient lists to identify patients with MbC or unresect-able locally advanced breast cancer. Patient demographic, clin-ical, pathologic, and outcome data were gathered from eMrs and coded to protect patient privacy. We selected subjects who had received capecitabine as a single agent or with trastuzumab for Her2-positive cases, as the first, second, or third line of che-motherapy in the metastatic setting from January 2006 to March 2011. Our decision to include patients treated with trastuzumab reflects the fact that the use of trastuzumab is now widely accept-ed as the standard of care for patients with Her2-positive disease, along with the fact that our comparator studies did not exclude trastuzumab-treated patients. Patients who received capecitabine with lapatinib were excluded because lapatinib treatment re-quires a modified starting dose of capecitabine and was not rep-resented in the comparator studies. Patients with measurable or nonmeasurable disease were eligible, although only patients with measurable disease on the basis of radiology reports and physical examination were included in the comparison of outcomes with published trials that used standard fDa-approved dosing.
Outcome AssessmentsOur study was conducted as a retrospective cohort analysis of the medical records of eligible patients. The primary endpoint was progression-free survival (PfS), defined as the duration in time between date capecitabine treatment started and date of progres-sion, with patients who did not have progression censored at the last follow-up date. We followed the principle of intent to treat (ITT), in that patients were not censored because of deviation from the intended capecitabine treatment. Progression was de-fined clinically and based upon evidence from radiology reports and physical examination. Included in this definition of progres-sion was the decision by the treating physician to initiate palliative radiation therapy, due to the fact that such decisions often reflect refractoriness to the current chemotherapy regimen and overall dis-ease progression. formal tumor assessment using the response evaluation Criteria in Solid Tumors (reCIST) was not done.
The primary aim of this study was a comparison of median PfS for uSC patients with measurable disease with the report-ed median PfS in published trials that used standard fDa-ap-proved dosing. Our major secondary aim was a comparison of PfS between patients in this cohort who had measurable disease and those who had nonmeasurable disease. additional analyses were performed to explore the relationship between PfS and various clinical and tumor characteristics in all patients (both patients with and without measurable disease).

22 www.ajho.com february 2015
· breast cancer ·
Statistical Analysisanalysis of PfS in relation to patient and disease characteris-tics was based on the log rank test, product-limit estimator, and univariate and multivariate Cox regression analysis.21 Patient or disease characteristic variables were each examined for their re-lationship with PfS, adjusting for whether or not a patient had measurable disease. Variables examined included treating site (LaC+uSC Medical Center or Norris Comprehensive Cancer Center), race/ethnicity, age at initial diagnosis of breast cancer, age at initiation of capecitabine treatment, biomarker status (es-trogen receptor [er], progesterone receptor [Pr], and Her2), prior treatment with anthracycline or taxane, line of adminis-tration of capecitabine therapy (1, 2, or 3), disease-free interval (DfI; defined as time between initial histological diagnosis of breast cancer and diagnosis of metastatic disease), concurrent administration of trastuzumab with capecitabine, the num-ber of metastatic disease sites, presence of visceral disease, and capecitabine dose per bSa. The variables that were associated with PfS at P ≤.20 after controlling for measurable disease status were then included in a multivariate Cox regression model, and a backward stepwise model selection method was used to select a final model by successively dropping nonsignificant variables from the model and refitting reduced models until all remaining variables were statistically significant at 0.20.22 Statistical analy-ses were performed using Stata Statistical Software (version 11.0; StataCorp LP, College Station, TX).
ResultsPatient CharacteristicsWe identified a total of 84 eligible patients who were treated with capecitabine alone or with trastuzumab between January 2006 and March 2011. Patient and disease characteristics are shown in Table 1. With the exception of the ethnic composition, baseline demographics and prognostic indicators including age, receptor status, DfI, and prior therapies were comparable between both hospital cohorts and those in the 2 major randomized controlled trials19,20 that serve as our comparators. Notably, 49% of patients were Hispanic, reflecting the large proportion of patients of this ethnicity seen at the uSC hospitals. Sixty-two (74%) patients had measurable disease and 16 (19%) had nonmeasurable disease. for the remaining 6 (7%) patients, measurability could not be ascertained. Twenty-four percent of patients had Her2-positive disease; 75% of these patients received trastuzumab concurrent-ly. Of the Her2-negative cases, 76% had hormone receptor (Hr; er or Pr) –positive tumors and 24% had triple-negative tumors. Seventy-five percent of patients had received some prior chemo-therapy in any setting before receiving capecitabine, while 25% of patients were chemotherapy-naïve. Overall, 70% of patients had been pretreated with an anthracycline, a taxane, or both. The majority of patients in our study (77%) received capecitabine as the first line; 17% received capecitabine as second-line, and 6% of patients received capecitabine as the third-line chemotherapy in the metastatic setting. Overall, our cohort was well matched to those in the chosen phase 3 comparator studies.
Capecitabine Exposureall 84 patients in our study received capecitabine at dosages sig-nificantly lower than the dosage approved by the fDa (1250 mg/m2 twice daily) or reported in the literature.8-10,12 Our use of flat dosing, rather than according to bSa, introduced some hetero-geneity in dosage per bSa received by the patients in this cohort. The median starting dosage was 565 mg/m2 twice daily, with a standard deviation of 115 mg/m2 twice daily and a range of 305 mg/m2 to 1057 mg/m2 twice daily (Figure 1). Since we typically used a flat-dosing method, the majority of our patients (n = 72; 86%) received an absolute dosage of 1000 mg twice daily. Of the remaining 12 patients, 1 (1%) received 500 mg twice daily, 4 (5%) received 750 mg twice daily, and 7 (8%) received 1500 mg twice daily. The median absolute dosage was 1017 mg twice daily, with a standard deviation of 163 mg twice daily. Two-thirds of our patients received capecitabine at dosages under 600 mg/m2
twice daily, less than half of the fDa-approved dosage of 1250 mg/m2 twice daily.
Efficacy of Capecitabine TherapyOf the 84 patients in our study, a total of 64 patients had disease progression during capecitabine treatment, including 52 patients who progressed on capecitabine as determined clinically by the
70
60
50
40
30
20
10
0300 400 500 600 700
Dose twice daily (miligrams) per body surface area
Perc
ent o
f Pat
ient
s
800 900 1000 1100
figure 1. Distribution of Capecitabine Dosage Per Body Surface Area
Distribution of capecitabine dosage per body surface area in this cohort. Range, 305-1057 mg/m2 twice daily; median dosage, 565 mg/m2 twice daily; standard deviation = 115 mg/m2 twice daily BSA indicates body surface area; PFS, progression-free survival.

VOL. 11, NO. 2 THe aMerICaN JOurNaL Of HeMaTOLOGy/ONCOLOGy 23
Very-Low-Dose CapeCitabine in MbC
Table 1. Patient and Disease Characteristics
Variable Total (N = 84) %
Measurable Disease
No 16 21%
Yes 62 79%
Unknown 6
Hospital
Los Angeles County Hospital (LAC+USC)
46 55%
Norris Comprehensive Cancer Center
38 45%
Race/Ethnicity
Hispanic 39 49%
White (non-Hispanic) 31 39%
Asian/Pacific Islander 6 7%
African/African American 4 5%
Unknown 4
Age at Diagnosis of Breast Cancer
Mean, median (range) 49 years, 48 years (22-85 years)
<50 years 47 56%
≥50 years 37 44%
Age at Initiation of Capecitabine
Mean, median (range) 55 years, 55 years (24-88 years)
<50 years 36 43%
≥50 years 48 57%
Tumor Biomarkers Subtype
HER 2-, ER+ or PR + 49 58%
HER 2-, ER -, PR - 15 18%
HER 2+ 20 24%
Prior Chemotherapy (neoadjuvant, adjuvant, or metastatic setting)
No 21 25%
Yes 63 75%
Pretreatment With Anthracyline/Taxane
Anthracycline only 9 11%
Taxane only 9 11%
Both 39 48%
Neither 25 30%
Unknown 2 31%
Prior Hormonal Therapy (adjuvant or metastatic setting)
No 26
Yes 58 69%
BSA indicates body surface area; ER, estrogen receptor; HER2, hu-man epidermal growth factor receptor 2; PR, progesterone receptor.
Variable Total (N = 84) %
Disease-Free Interval
Mean, median (range) 3.9 years, 2.6 years (0-14.8 years)
0 (stage IV disease at presentation) 16 19%
<1 year 6 7%
≥1 year and <2 years 11 13%
≥2 years and <5 years 23 28%
>5 years 28 33%
Capecitabine: Line of Chemotherapy in Metastatic Setting
1 65 77%
2 14 17%
3 5 6%
Trastuzumab Administered With Capecitabine
No 68 82%
Yes 15 18%
Unknown 1
Number of Disease Sites at Start of Capecitabine Therapy
1-2 42 51%
>2 40 49%
Unknown 2
Visceral Disease
No 27 33%
Yes 56 67%
Unknown 1
Brain Metastases
No 74 89%
Yes 9 11%
Unknown 1
Bone Disease Only
No 74 89%
Yes 9 11%
Unknown 1
Body Surface Area (BSA)
Mean, median (range) 1.8 m2, 1.8 m2 (1.3 m2-2.4 m2)

24 www.ajho.com february 2015
· breast cancer ·
treating physician, 7 patients who commenced radiation therapy while taking capecitabine, 2 patients who progressed after fin-ishing capecitabine with no intervening antineoplastic therapy, and 4 patients who progressed on the subsequent therapy, as we used an ITT approach with progression as the desired outcome variable. Of these 4 patients, 2 discontinued capecitabine due to toxicity. Of the 20 patients who did not have disease progression, 10 patients did not progress as of the end of our study period and 10 were lost to follow-up. Median follow-up in these 20 patients was 5.3 months (range, 0.5-56 months). baseline patient and tu-mor characteristics are shown in Table 1.
Since the 2 major published randomized trials examining the efficacy of single-agent capecitabine both restricted their analy-ses of PfS to patients with measurable disease,19,20 we also deter-mined median PfS for patients in our cohort with measurable disease. Median PfS for patients with measurable disease was 4.1 months (95% confidence interval [CI], 2.9-5.7; Figure 2A, dashed line), which is similar to the median PfS values of 4.4 months (95% CI, 4.1-5.4; n = 480)19 and 4.2 months (95% CI, 3.8-4.5; n = 377)20 previously reported. a total of 55 patients with measurable disease received flat dosages of capecitabine of 1000 mg twice daily. Median PfS in these patients was 4.1 months (95% CI, 2.8-5.7 months), which was almost identical to the me-dian PfS for all patients with measurable disease.
Since a secondary aim was to compare PfS between patients in this cohort with and without measurable disease, we also de-termined PfS for patients with nonmeasurable disease. These patients had a significantly longer PfS than patients with mea-surable disease (19.7 months; 95% CI, 3.9-28.5; Figure 2A, solid line).
The relationship between median PfS and each patient and disease characteristic variable was examined, and the hazard ra-tios for progression for each variable, with associated 95% CI, are shown in Figure 2B.
for the remaining analyses, we included both measurable and nonmeasurable cases. Patients were stratified based on measur-able disease status, and the relationship between PfS and each patient and disease characteristic variable known to influence disease progression was examined. Stratifying by measurable disease status, univariate analyses showed that factors associat-ed with PfS at P ≤.05 included biomarker status, DFI, age at initiation of capecitabine therapy, and prior hormonal therapy. Specifically, triple-negative (er-, Pr-, Her2-) status was associat-ed with a lower PfS, as was shorter DfI. Concurrent administra-tion of trastuzumab produced a trend toward increased PfS (P = .12). Patients with measurable disease who did not receive trastu-zumab concurrently with capecitabine had a median PfS of 3.7 months (95% CI, 2.8-5.7; n = 51). Multivariate Cox regression analyses showed that besides measurable disease status, biomark-er status and DfI were significantly associated with PfS at P ≤.05 independent from other factors, and receiving trastuzumab con-
currently with capecitabine was marginally associated with PfS (P = .077). Odds ratios for progression based on these factors are shown in Table 2. Kaplan-Meier curves for these independently associated factors are depicted in Figure 3.
Demographic factors not independently associated with PfS at the significance level of 0.20 included race and age at initial diagnosis of breast cancer. Notably, within the range of low dos-ages administered to this cohort, dosage of capecitabine was not independently associated with PfS at P = .20, with all three dos-age tertiles demonstrating similar median PfS (Figure 4). Other treatment-related factors not independently associated with PfS at P = .20 included history of administration of any prior chemo-
0.8
0.9
1.0
0.7
0.6
0.5
0.3
0.4
0.2
0.1
0.00 1 2 3
Times in years from start of capecitabine treatment
PFS
Prob
abili
ty
4 5
No Measurable Disease Median PFS 19.7 m (95%CI 3.9~28.5)(n = 16)Median PFS 4.1 m (95%CI 2.9~5.7)(n = 62)P = .029
Measurable Disease
figure 2. Kaplan-Meier Curve Showing PFS
Kaplan-Meier curve showing PFS for patients with and without measurable disease; (B) PFS hazard ratios and associated 95% CIs for subset analyses. BSA indicates body surface area; DFI, disease-free interval; ER, estrogen receptor; HER2, human epidermal growth factor receptor 2; PR, progesterone receptor.
(A)
(B)
Hazard Ratio and 95% CI
0.1 0.5 1 2 5 10
Measurable vs Non-Measurable
Capecitabine Dose per BSA: 3rd Tertile vs 1st
Capecitabine Dose per BSA: 2nd Terile vs 1st
Brain Mets: Yes vs No
Disease Sites: >2 vs <=2
Trastuzumab with Capecitabine: Yes vs No
Capecitabine 2nd or 3rd Line vs 1st Line
DFI >=5 y vs 0 y
DFI >0 y and <5 y vs 0 y
Prior Homonal Therapy: Yes vs No
Anthracyline/Taxane: Either vs Neither
Prior Chemo Yes vs No
Her2+ vs Her2- (ER+ or PR+)
Her2-, ER-, PR- vs Her2- (ER+ or PR+)
Age at Start of Capecitabine: >=50 vs <50 y
Norris Cancer Center vs LA County Hosp.Other/Unknown Race vs Hispanic
White vs Hispanic
Visceral Disease: Yes vs No

VOL. 11, NO. 2 THe aMerICaN JOurNaL Of HeMaTOLOGy/ONCOLOGy 25
Very-Low-Dose CapeCitabine in MbC
therapy, history of administration of anthracyclines or taxanes, and line of therapy of capecitabine in the metastatic setting (1, 2, or 3; Figures 5A, B, and C). The median PfS was 3.7 months (95% CI, 2.5-5.5; n = 46) for patients with measurable disease who were previously treated with either anthracyclines or tax-anes, which was similar to the PfS value of 4.1 months deter-mined for all patients with measurable disease.
finally, for the 4 patients who progressed on the next therapy after capecitabine was terminated without progression, censor-ing these patients at the time capecitabine was stopped did not alter the median PfS among patients with measurable disease.
DiscussionOur retrospective analysis suggests that the administration of low starting dosages of capecitabine is as effective in the treatment of MbC as the full fDa-approved dosage. although it is well established that the fDa-approved dosage of capecitabine is as-sociated with frequent and treatment-limiting toxicities, little or no efficacy data from phase 3 randomized, controlled trials exist to support the administration of capecitabine at lower dosages.
Here, we report data demonstrating efficacy of capecitabine in MbC at significantly lower starting dosages (median dosage of 565 mg/m2 twice daily, or 1130 mg/m2 daily) than any values previously reported in the literature.7-13 We first presented our re-sults at the 2012 meeting of the american Society of Clinical On-cology.23 Since that time, ambros et al24 have published a similar study that replicated our results. This study was a retrospective analysis of 86 patients treated at a single breast-focused oncology practice with a fixed low dosage of capecitabine monotherapy for Her2-negative MbC. The authors concluded that a flat dosage of 1000 mg twice daily (median starting dosage of 633.5 mg/m2 twice daily, or about half the fDa-approved dosage of 1250 mg/m2 twice daily) had a similar clinical benefit rate, as determined by objective response rate and TTP, to the full dosage based on a historical comparison of 12 prior trials involving 1949 patients. These results reinforce the results of our study and strengthen our recommendation for a prospective randomized, controlled trial evaluating the efficacy of low dosages of capecitabine as sin-gle-agent therapy, or in combinations that have shown benefit over single-agent therapy.
Since patients typically received a flat dosage of capecitabine of 1000 mg twice daily, there was some heterogeneity of dosage per bSa within this cohort. However, we observed no differences in PfS between any of the 3 strata of dosages per bSa, which lends support to the evidence that precise dosing based on bSa may be unnecessary, as has been suggested by others.25 furthermore, only 2 patients in our study (2.4%) discontinued capecitabine due to toxicity. Previous clinical trials reported rates of discon-tinuation due to toxicity of 11%20 and 11.9%26 for capecitabine dosages of 1250 mg/m2 and 1000 mg/m2 twice daily, respec-tively. Though this analysis does not allow for formal statistical
Median PFS 8.5 m (n = 49)Median PFS 3.0 m (n = 15)Median PFS 6.0 m (n = 20)Unstratified: P = .003Stratified: P = .004
Her2-, ER or PR+Triple NegativeHer2+
Time in years from start of capecitabine treatment
PFS
Prob
abili
ty
0.8
0.9
1.0
0.7
0.6
0.5
0.3
0.4
0.2
0.1
0.00 1 2 3 4 5
figure 3. Factors That Significantly Influenced PFS
Factors that significantly influenced PFS on capecitabine therapy in patients with and without measurable disease. (A) tumor biomarker status; (B) DFI; and (C) concurrent trastuzumab ad-ministration. For all figures, unstratified P value refers to P value in univariate analysis; stratified P value refers to P value after controlling for measurable disease status. DFI indicates disease-free interval; ER, estrogen receptor; HER2, human epidermal growth factor receptor 2; PFS, progression-free survival; PR, progesterone receptor.
Median PFS 3.0 m (n = 16)Median PFS 5.2 m (n = 40)Median PFS 8.5 m (n = 20)Unstratified: P = .024Stratified: P = .043
DFI = 0 y0< DFI <5 yDFI >=5 y
Time in years from start of capecitabine treatment
PFS
Prob
abili
ty
0.8
0.9
1.0
0.7
0.6
0.5
0.3
0.4
0.2
0.1
0.00 1 2 3 4 5
Time in years from start of capecitabine treatment
PFS
Prob
abili
ty
Median PFS 4.1 m (n = 68)Median PFS 9.6 m (n = 15)Unstratified: P = .20Stratified: P = .12
No Trastuzumab with CapecitabineTrastuumab with Capecitabine
0.8
0.9
1.0
0.7
0.6
0.5
0.3
0.4
0.2
0.1
0.00 1 2 3 4 5
(A)
(B)
(C)
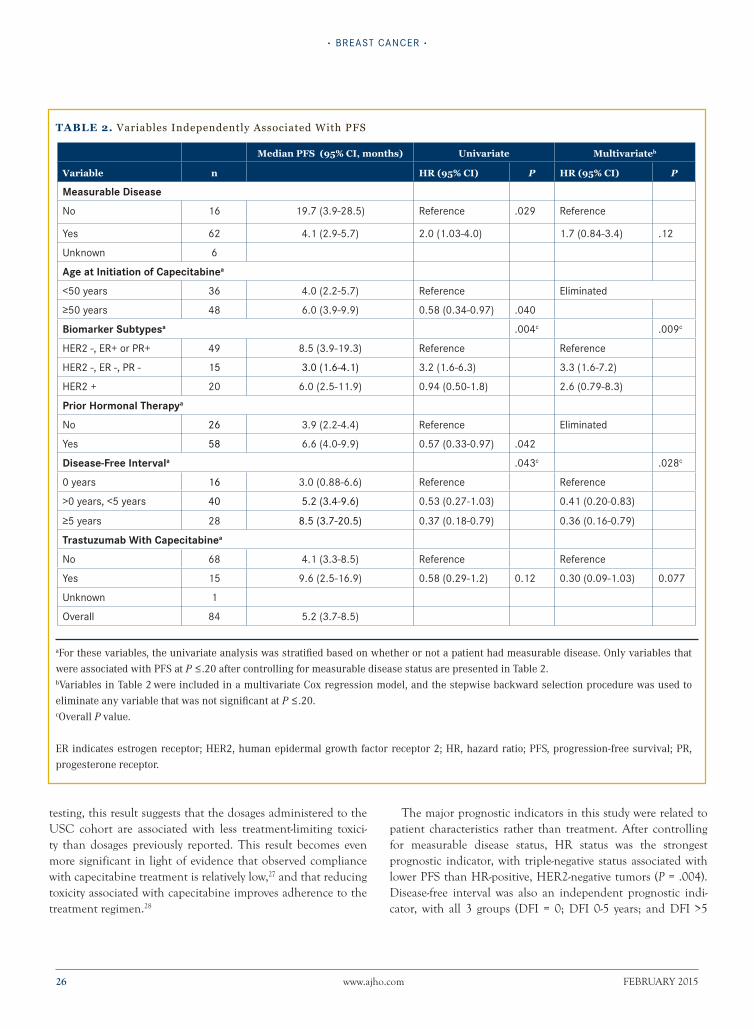
26 www.ajho.com february 2015
· breast cancer ·
testing, this result suggests that the dosages administered to the uSC cohort are associated with less treatment-limiting toxici-ty than dosages previously reported. This result becomes even more significant in light of evidence that observed compliance with capecitabine treatment is relatively low,27 and that reducing toxicity associated with capecitabine improves adherence to the treatment regimen.28
The major prognostic indicators in this study were related to patient characteristics rather than treatment. after controlling for measurable disease status, Hr status was the strongest prognostic indicator, with triple-negative status associated with lower PfS than Hr-positive, Her2-negative tumors (P = .004). Disease-free interval was also an independent prognostic indi-cator, with all 3 groups (DfI = 0; DfI 0-5 years; and DfI >5
Table 2. Variables Independently Associated With PFS
Median PfS (95% Ci, months) univariate Multivariateb
Variable n Hr (95% Ci) P Hr (95% Ci) P
Measurable Disease
No 16 19.7 (3.9-28.5) Reference .029 Reference
Yes 62 4.1 (2.9-5.7) 2.0 (1.03-4.0) 1.7 (0.84-3.4) .12
Unknown 6
Age at Initiation of Capecitabinea
<50 years 36 4.0 (2.2-5.7) Reference Eliminated
≥50 years 48 6.0 (3.9-9.9) 0.58 (0.34-0.97) .040
Biomarker Subtypesa .004c .009c
HER2 -, ER+ or PR+ 49 8.5 (3.9-19.3) Reference Reference
HER2 -, ER -, PR - 15 3.0 (1.6-4.1) 3.2 (1.6-6.3) 3.3 (1.6-7.2)
HER2 + 20 6.0 (2.5-11.9) 0.94 (0.50-1.8) 2.6 (0.79-8.3)
Prior Hormonal Therapya
No 26 3.9 (2.2-4.4) Reference Eliminated
Yes 58 6.6 (4.0-9.9) 0.57 (0.33-0.97) .042
Disease-Free Intervala .043c .028c
0 years 16 3.0 (0.88-6.6) Reference Reference
>0 years, <5 years 40 5.2 (3.4-9.6) 0.53 (0.27-1.03) 0.41 (0.20-0.83)
≥5 years 28 8.5 (3.7-20.5) 0.37 (0.18-0.79) 0.36 (0.16-0.79)
Trastuzumab With Capecitabinea
No 68 4.1 (3.3-8.5) Reference Reference
Yes 15 9.6 (2.5-16.9) 0.58 (0.29-1.2) 0.12 0.30 (0.09-1.03) 0.077
Unknown 1
Overall 84 5.2 (3.7-8.5)
aFor these variables, the univariate analysis was stratified based on whether or not a patient had measurable disease. Only variables that
were associated with PFS at P ≤.20 after controlling for measurable disease status are presented in Table 2. bVariables in Table 2 were included in a multivariate Cox regression model, and the stepwise backward selection procedure was used to
eliminate any variable that was not significant at P ≤.20. cOverall P value.
ER indicates estrogen receptor; HER2, human epidermal growth factor receptor 2; HR, hazard ratio; PFS, progression-free survival; PR,
progesterone receptor.

VOL. 11, NO. 2 THe aMerICaN JOurNaL Of HeMaTOLOGy/ONCOLOGy 27
Very-Low-Dose CapeCitabine in MbC
years) associated with statistically significant differences in PfS (P = .043). These findings are consistent with prior studies re-garding prognostic indicators in MbC.29 Several patient-related variables typically associated with lower PfS, including more than 2 disease sites, presence of visceral disease, and presence of central nervous system disease, were not found to be significant prognostic indicators in our analysis.
The initial trials leading to the approval of capecitabine en-rolled patients defined as resistant to anthracycline3 or taxane4 chemotherapy, as did our comparator studies,19,20 which com-pared the efficacy of capecitabine alone against capecitabine plus ixabepilone. Other trials have examined the efficacy of capecit-abine as first-line chemotherapy for MbC, demonstrating superi-ority over cyclophosphamide, methotrexate, and 5-fluorouracil, albeit with respect to OS and not PfS in the case of the larger trial,13 possibly due to post-progression therapy.13,29 These find-ings have helped to establish first-line capecitabine therapy as a standard of care in MbC. Our study reflects these evolving pat-terns of capecitabine administration, including 77% of patients who received capecitabine as the first line and 23% who received capecitabine as second- or third-line chemotherapy in the meta-static setting. Notably, neither line of treatment of capecitabine therapy nor prior administration of anthracyclines or taxanes were significant predictors of PfS, suggesting that low starting dosages of capecitabine are efficacious both as first-line chemo-therapy and in patients who have been more heavily pretreated in the metastatic setting.
Our results support the equal efficacy of low-dose capecitabine among patients with measurable disease, but also suggest that the responses of patients without measurable disease may be fundamentally different. To maximize comparability with previ-ous studies, patients without measurable disease were excluded from our primary analysis of PfS. However, we determined that measurability of disease was a significant predictor of PfS for patients in our cohort, as patients with measurable disease had a median PfS of 4.1 months and patients without measurable disease had a median PfS of 19.7 months. for various reasons, patients without measurable disease have typically been excluded from analysis in clinical trials, and as a result, less information is available regarding their responses to treatment. Nevertheless, these patients represent a sizeable, and thus clinically important, subset of patients with MbC. a large proportion of our patients (and MbC patients in general) without measurable disease had metastases in bone only, and it is known that bone-only tumors are more likely to be Hr-positive and possess other less aggres-sive tumor characteristics.30,31 additionally, there is evidence that patients with bone-only disease or disease that is otherwise not measurable may be a biologically distinct subgroup of patients, and therefore should be analyzed separately.32 Our study empha-sizes the need for further research on patients without measur-able disease in order to facilitate the delivery of informed, evi-
dence-based care for this subset of patients.Our results also provide preliminary data on responses to
capecitabine in a cohort with a significant proportion of Hispan-ic patients. Little, if any, of the prior work on capecitabine has re-ported data on responses among Hispanic patients, but there is evidence that disease characteristics and responses to treatment may be different in Hispanics than in other ethnic groups.33,34 Though we did not observe a difference in PfS on capecitabine therapy between Hispanic and non-Hispanic patients, we were unable to gather data on the frequency of various treatment-relat-ed aes. It is possible that pharmacogenomics and other factors differ between patients of different ethnicities, and more studies in ethnic subsets are needed.
This study presents evidence that capecitabine is effective at significantly lower starting dosages than previously appreciated, and supports decisions by clinicians to initiate treatment at lower dosages than the one approved by the fDa. The main limitation of our study is its retrospective nature. as such, we were unable to collect information on dosage reductions or the frequency of various drug toxicities. While this cohort was heterogeneous in lines of therapy and prior treatments received, these factors were not significant predictors of PfS. additionally, although we were able to quantify PfS, we were unable to use formal reCIST cri-teria to evaluate tumor responses, and as a result were unable to separate the nonprogressors into complete responders, partial responders, and stable disease, and to perform a more in-depth analysis based on those parameters. for these reasons, a random-
Time in years from start of capecitabine treatment
PFS
Prob
abili
ty
Median PFS 4.1 m (n = 27)Median PFS 5.2 m (n = 28)Median PFS 5.4 m (n = 28)Unstratified: P = .20Stratified: P = .34
Dose per BSA: 1st tertileDose per BSA: 2nd tertileDose per BSA: 3rd tertile
0.8
0.9
1.0
0.7
0.6
0.5
0.3
0.4
0.2
0.1
0.00 1 2 3 4 5
figure 4. PFS For Patients In Each Dosage Tertile
PFS for patients in each dosage tertile. 1st tertile: 305-536 mg/m2; 2nd tertile: 536.5-593 mg/m2; 3rd tertile: >593 mg/m2
(doses administered twice daily).
BSA indicates body surface area; PFS, progression-free survival.
28 www.ajho.com february 2015
· breast cancer ·
ized phase 3 trial examining capecitabine dosing is necessary in order to confirm these results.
ConclusionsIn the 13 years since the approval of capecitabine by the fDa, there has been mounting evidence that lowering the starting dos-age can reduce drug-associated toxicity without compromising antitumor efficacy.7-9,11,26 Our report adds to this evidence, and along with a subsequent report24 supports a prospective phase 3 randomized clinical trial of the fDa-approved dosage of capecit-abine against one or several lower starting dosages, coupled with appropriate pharmacogenomics studies, in order to optimize the benefit-to-toxicity ratio of this agent. future studies should also address the question of dosing method, as our results suggest that similar outcomes can be achieved regardless of whether pa-tients are given flat dosages or dosed based on bSa. updating the guidelines governing the use of capecitabine based on more definitive studies would have important implications for clini-cians and patients, as a milder toxicity profile may improve com-pliance by patients and lead to longer disease control with fewer discontinuations due to toxicity.
Affiliations: Caitlin bertelsen, MD, is from uSC Keck School of Medicine, Department of Otolaryngology/Head and Neck Surgery; Lingyun Ji, MS, and richard Sposto, PhD, are from the Department of Preventive Medicine and uSC/Norris Compre-hensive Cancer Center; agustin a. Garcia, MD, Christy russell, MD, and Darcy Spicer, MD, are from the Department of Medi-cine and the uSC/Norris Comprehensive Cancer Center; and Debu Tripathy, MD, is from The university of Texas MD ander-son Cancer Center, Department of breast Medical Oncology, Houston, TX, and is editor-in-chief of AJHO.Disclosures: Dr russell reports receiving honoraria from roche Pharmaceuticals as a member of the Speakers’ bureau. Dr Trip-athy has received a clinical trial contract with roche/Genentech (paid to academic institution). Drs bertelsen, Ji, Garcia, Spicer, and Sposto report no relevant financial conflicts of interest to disclose.Acknowledgment of funding: research was supported by the Keck School of Medicine Medical Student Summer research fellowship.Address correspondence to: Debu Tripathy, MD, university of Texas MD anderson Cancer Center, 1515 Holcombe, unit 1354, Houston, TX 77030. Phone: 733-792-2817; fax: 713-563-0903; email: [email protected].
ReFeRenCes1. Siegel r, Naishadham D, Jemal a. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11-30. 2. Cardoso f, bedard PL, Winer eP, et. al. International
Time in years from start of capecitabine treatment
PFS
Prob
abili
ty
Median PFS 8.7 m (n = 21)Median PFS 4.3 m (n = 63)Unstratified: P = .33Stratified: P = .60
No Previous ChemoWith Previous Chemo
0.8
0.9
1.0
0.7
0.6
0.5
0.3
0.4
0.2
0.1
0.00 1 2 3 4 5
figure 5. Treatment-Related Factors Not Signifi-cantly Associated With PFS
Treatment-related factors not significantly associated with PFS on capecitabine therapy. (A) History of any prior chemotherapy; (B) prior administration of anthracycline or taxane therapy; and (C) line of therapy of capecitabine in the metastatic setting.
PFS indicates progression-free survival.
Time in years from start of capecitabine treatment
PFS
Prob
abili
ty
Median PFS 8.5 m (n = 25)Median PFS 4.3 m (n = 57)Unstratified: P = .48Stratified: P = .78
No Prior Anthra or TaxaneReceived Prior Anthra or Taxane
0.8
0.9
1.0
0.7
0.6
0.5
0.3
0.4
0.2
0.1
0.00 1 2 3 4 5
Time in years from start of capecitabine treatment
PFS
Prob
abili
ty
Median PFS 5.2 m (n = 65)Median PFS 5.4 m (n = 19)Unstratified: P = .23Stratified: P = .55
1st Line2nd or 3rd
0.8
0.9
1.0
0.7
0.6
0.5
0.3
0.4
0.2
0.1
0.00 1 2 3 4 5
(A)
(B)
(C)

VOL. 11, NO. 2 THe aMerICaN JOurNaL Of HeMaTOLOGy/ONCOLOGy 29
Very-Low-Dose CapeCitabine in MbC
Guidelines for Management of Metastatic breast Cancer: Combination vs Sequential Single-agent Chemotherapy. J Natl Cancer Inst. 2009;101:1174-1181.3. Wagstaff a, Ibbotson T, Goa KL. Capecitabine: a review of its pharmacology and therapeutic efficacy in the management of advanced breast cancer. Drugs. 2003;63:217-236.4. O’Shaughnessy J, Miles D, Vukelja S, et. al. Superior survival with capecitabine plus docetaxel combination therapy in anthracycline-pretreated patients with advanced breast cancer: phase III trial results. J Clin Oncol. 2002;20:2812-2823. 5. blum JL, Jones Se, buzdar au, et. al. Multicenter phase II study of capecitabine in paclitaxel-refractory metastatic breast cancer. J Clin Oncol. 1999;17:485-493.6. Tripathy D. Capecitabine in combination with novel targeted agents in the management of metastatic breast cancer: underlying rationale and results of clinical trials. Oncologist. 2007;12:375-389. 7. Hennessy bT, Gauthier aM, Michaud Lb, et al. Lower dose capecitabine has a more favorable therapeutic index in metastatic breast cancer: retrospective analysis of patients treated at M.D. anderson Cancer Center and a review of capecitabine toxicity in the literature. Ann Oncol. 2005;16:1289-1296.8. Leonard r, Hennnessy bT, blum JL, et al. Dose-adjusting capecitabine minimizes adverse effects while maintaining efficacy: a retrospective review of capecitabine for metastatic breast cancer. Clin Breast Cancer. 2011;11:349-356.9. Kaufmann M, Maass N, Costa SD, et al. first-line therapy with moderate dose capecitabine in metastatic breast cancer is safe and active: results of the MONICa trial. Eur J Cancer. 2010;46:3184-3191. 10. rossi D, alessandroni P, Catalano V, et al. Safety profile and activity of lower capecitabine dose in patients with metastatic breast cancer. Clin Breast Cancer. 2007;7:857-860.11. Naughton M. evolution of capecitabine dosing in breast cancer. Clin Breast Cancer. 2010;10:130-135.12. Taguchi T, Nakayama T, Masuda N, et al. Study of low-dose capecitabine monotherapy for metastatic breast cancer. Chemotherapy. 2010;56:166-170. 13. Stockler M, Harvey VJ, francis Pa, et al. Capecitabine versus classical cyclophosphamide, methotrexate, and fluorouracil as first-line chemotherapy for advanced breast cancer. J Clin Oncol. 2011;29:4498-4504. 14. Traina Ta, Theodoulou M, feigin K, et al. Phase I study of a novel capecitabine schedule based on the Norton-Simon mathematical model in patients with metastatic breast cancer. J Clin Oncol. 2008;26(11):1797-1802.15. Gajria D, Gonzalez J, feigin K, et al. Phase II trial of a novel capecitabine dosing schedule in combination with lapatinib for the treatment of patients with Her2-positive metastatic breast cancer. Breast Cancer Res Treat. 2012;131:111-116.16. Gajria D, feigin K, Tan LK, et al. Phase 2 trial of a novel
capecitabine dosing schedule in combination with bevacizumab for patients with metastatic breast cancer. Cancer. 2011;117:4125-4131.17. Largillier r, etienne-Grimaldi MC, formento J, et al. Pharmacogenetics of capecitabine in advanced breast cancer patients. Clin Cancer Res. 2006;12:5496-5502.18. Midgley r, Katz, D. Capecitabine: have we got the dose right? Nat Clin Pract Oncol. 2009;6:17-24.19. Thomas eS, Gomez HL, Li rK, et al. Ixabepilone plus capecitabine for metastatic breast cancer progressing after anthracycline and taxane treatment. J Clin Oncol. 2008;25:5210-5217.20. Sparano Ja, Vrdoljak e, rixe O, et al. randomized phase III trial of ixabepilone plus capecitabine versus capecitabine in patients with metastatic breast cancer previously treated with an anthracycline and a taxane. J Clin Oncol. 2010;28:3256-3263. 21. Cox Dr, Oakes D. Analysis of Survival Data. New york: Chapman and Hall; 1984. 22. Hocking rr. The analysis and selection of variables in linear regression. Biometrics. 1976;32:1-49.23. bertelsen C, Ji L, Garcia aa, et al. efficacy of low-dose capecitabine in metastatic breast cancer. J Clin Oncol. 2012;30(suppl; abstr 1065).24. ambros T, Zeichner Sb, Zaravinos J, et al. a retrospective study evaluating a fixed low dose capecitabine monotherapy in women with Her-2 negative metastatic breast cancer. Breast Cancer Res Treat. 2014;146:7-14.25. Partridge a, archer L, Kornblith ab, et al. adherence and persistence with oral adjuvant chemotherapy in older women with early-stage breast cancer in CaLGb 49907: adherence companion study 60104. J Clin Oncol. 2010;28:2418-2422. 26. robert NJ, Dieras V, Glaspy J, et al. rIbbON-1: randomized, double-blind, placebo-controlled phase III trial of chemotherapy with or without bevacizumab for first-line treatment of human epidermal growth factor receptor 2-negative, locally recurrent or metastatic breast cancer. J Clin Oncol. 2011;29:1252-1260.27. Winterhalder r, Hoesli P, Delmore G, et al. Self-reported compliance with capecitabine: findings from a prospective cohort analysis. Oncology. 2011;80:29-33.28. Gilabert M, bertucci f, esterni b, et al. Capecitabine after anthracycline and taxane exposure in Her2-negative metastatic breast cancer patients: response, survival and prognostic factors. Anticancer Res. 2011;31:1079-1086.29. O’Shaughnessy J, blum JL, Moiseyenko V, et al. randomized, open-label phase II trial of oral capecitabine (Xeloda) vs. a reference arm of intravenous CMf (cyclophosphamide, methotrexate, and 5-fluorouracil) as first-line therapy for advanced/metastatic breast cancer. Ann Oncol. 2001;12:1247-1254.30. Gupta GP, Minn aJ, Kang y, et al. Identifying site-specific metastasis genes and functions. Cold Spring Harb Symp Quant Biol. 2005;70:149-158.

30 www.ajho.com february 2015
· breast cancer ·
31. Minn aJ, Gupta GP, Siegel PM, et al. Genes that mediate breast cancer metastasis to lung. Nature. 2005;436:518-524.32. Kang y, Siegel PM, Shu W, et al. a multigenetic program mediating breast cancer metastasis to bone. Cancer Cell. 2003;3:537-549. 33. Patel T, Colon G, bueno Hume C, et al. breast cancer in Latinas: gene expression, differential response to treatments, and differential toxicities in Latinas compared with other population groups. Oncologist. 2010;15:466-475. 34. Watlington T, byers T, Mouchawar J, et al. Does having insurance affect differences in clinical presentation between Hispanic and non-Hispanic white women with breast cancer? Cancer. 2007;109:2093-2099.

VOL. 11, NO. 2 THE AMERICAN JOURNAL OF HEMATOLOGY/ONCOLOGY 31
· commentary ·
New Year’s Resolution: Work to Do
Omid Hamid, MD
As 2015 begins, it is with great anticipation that we await the results of ongoing work in the field of melanoma therapy. We have come a long way, as immunotherapy
with PD-1/PD-L1 checkpoint inhibition has gained approval as a standard therapy. Multiple phase 3 trials have expedient-ly reached accrual, and the results of these studies could thrust PD-1 axis blockade into the first-line setting as standard therapy. We have moved past single-agent targeted BRAF therapy, with improved progression-free and overall survival with combined BRAF/MEK inhibition.1,2 These are game changers; higher re-sponse rates and faster kinetics of response seen with the PD-1/PD-L1 checkpoint and BRAF/MEK inhibitors translate into an overall survival advantage. While the targeted agents may have higher response rates, we have not seen responses as durable as those seen with immunotherapy. In the past, this would be enough. Fortunately, today we realize that there is more. We have just begun to touch on the promise of immunotherapy. The future will elucidate optimal combinations of checkpoint inhibition and other immune-oncologic modalities and targeted agents. Answers will not only seek most favorable combinations but also appropriate dose, sequence, and schedule.
The field of checkpoint inhibition began with ipilimumab and progressed to PD-1/P-L1 inhibition. Today, there exist phase 1 trials testing novel inhibitory targets such as TIM-3 and LAG-3, with preclinical data proposing the benefit of combination therapy. Agonistic antibodies against T-cell co-stimulatory recep-tors (OX-40, GITR, CD137) have also burst onto the scene with multiple phase 1 trials in their early stages. These agents will look to replicate the success of their predecessors and will pro-vide further options for our patients. We now know that lack of response to one immune therapy does not preclude subsequent response to another.
Oncologists today have the mandate to reconfigure their pre-conceived notions of immunotherapy. The toolbox has expand-ed past just interleukin-2, interferon, and ipilimumab. Newer cytokines (IL12, IL21), vaccines, immune suppressive enzyme inhibitors (IDO inhibitors), adoptive cell transfer, oncolytic im-munotherapy, and T-cell engineering have expanded our arma-mentarium. The need to familiarize oneself with these options is tantamount. For example, the oncolytic therapy talimogene laherparepvec (T-VEC) utilizes an attenuated herpes virus that can only replicate in malignant cells. Unfettered viral replication
leads to focal cancer cell lysis while secreting GM-CSF, more virus capable of infecting other malignant cells, and tumor antigens creating an immune stimulatory environment. Today, T-VEC therapy has shown local and distant tumor response, improved response rates and tumor control, and a trend toward improved survival. The minimal side effects seen with this therapy have made it an ideal candidate for combination immunotherapy. It is no surprise that recent data in combination with ipilimumab have shown improved response rates, and clinical trials of T-VEC with PD-1 therapy will begin in the near future.
Combinations hold the potential to overcome multiple bar-riers that tumor cells possess to evade a host immune response and provide an overall survival benefit to a greater proportion of patients. Initial forays into combination therapies for melanoma resulted in toxicity and discouragement.3 This is not the current state of combination therapy. With the multitude of targeted therapies, immune modulators, and checkpoint inhibitors/ag-onists, extensive options exist. Early attempts in melanoma at combining radiotherapy and immunotherapeutics have shown promise of improvement in local and systemic control.4
Our mandate now emphasizes the importance of translating the advances made in the field toward the best patient outcomes. The major impetus toward this goal is based predominantly on predictive biomarkers that accurately foretell response to tumor immunotherapy. This dynamic paradigm holds the promise of appropriate patient selection and improved response rates while sparing patients valuable time and the expense of treatment-re-lated morbidity. Recent trials have focused on requirements of tumor specimens (both archival and fresh) in addition to blood samples at multiple time points in therapy, including response and progression, in an effort to unlock this holy grail. We have also begun to retrospectively look for these markers in established therapies, including CTLA-4 blockade.5 Initial data with PD-1/PD-L1 checkpoint inhibition indicating tumor PD-L1 staining as a go-no-go predictor was premature. PD-L1-negative tumors re-spond, but they do so at a lower rate, and therefore we continue to develop improved biomarkers. Wolchok et al6 have indicated that dual checkpoint blockade may offer the ability to increase response rates in this population. Newer techniques focusing on pre-existing CD8+ T cells distinctly located at the invasive tumor margin7 and PD-L1 expression by tumor-infiltrating immune cells8 have indicated a more sensitive predictive approach.
MELANOM
A
SPECIAL
SECTION

32 www.ajho.com FEBRUARY 2015
new year’s resolution: work to do
Led by the advances made in melanoma, immuno-oncology is burgeoning into a separate discipline amidst all cancers. This approach is actively investigated for its potential to translate from melanoma into similar breakthroughs in long-term survival in multiple tumor types. Currently, PD-1 and PD-L1 checkpoint inhibitors have garnered “breakthrough designation” in lung cancer, bladder cancer, and Hodgkin lymphoma. Checkpoint inhibition has already shown survival advantage versus chemo-therapy in both lung cancer9 and melanoma.10 As the field of immuno-oncology expands, so does our need to understand the best candidates for its benefit, and the most optimal combina-tions and sequences. We have only begun this journey. It is clear that today we have many more tools in the tool box, and there is still much work to do.
In the special melanoma section that follows, we will take a more in-depth look at some of these advances and what they may mean to the treatment of patients with melanoma.
Affiliation: Omid Hamid, MD, is director, Melanoma Program, and chief, Translational Research and Immuno-Oncology at The Angeles Clinic and Research Institute in Los Angeles, CA. Disclosure: Dr Hamid reports no relevant financial conflicts of interest to disclose. Address correspondence to: Omid Hamid, MD, The Angeles Clinic and Research Institute, 11818 Wilshire Blvd, Suite 200, Los Angeles, CA 90025. Phone: 310-231-2185.
RefeRences1. Larkin J, Ascierto PA, Dreno B, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014;371:1867-1876.2. Robert C, Karaszewska B, Schachter J, et al. Improved over-all survival in melanoma with combined dabrafenib and trame-tinib. N Engl J Med. 2015;372:30-39.3. Ribas A, Hodi FS, Callahan M, Konto C, Wolchok J. Hepa-totoxicity with combination of vemurafenib and ipilimumab. N Engl J Med. 2013;368:1365-1366.4. Postow MA, Callahan MK, Barker CA, et al. Immunologic correlates of the abscopal effect in a patient with melanoma. N Engl J Med. 2012;366(10):925-931.5. Snyder A, Makarov V, Merghoub T, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371(23):2189-2199. 6. Wolchok JD, Kluger H, Callahan MK, et al. Nivolum-ab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369(2):122-133.7. Tumeh PC, Harview CL, Yearley JH, et al. PD-1 blockade in-duces responses by inhibiting adaptive immune resistance. Na-ture. 2014;515(7528):568-571.8. Herbst RS, Soria JC, Kowanetz M, et al. Predictive correlates of response to anti-PD-L1 antibody MPDL3280A in cancer pa-
tients. Nature. 2014;515(7528):563-567.9. Study of BMS-936558 (Nivolumab) Compared to Docetaxel in Previously Treated Advanced or Metastatic Squamous Cell Non-small Cell Lung Cancer (NSCLC) (CheckMate 017). Clini-calTrials.gov website. NCT01642004.10. Robert C, Long GV, Brady B, et al. Nivolumab in previous-ly untreated melanoma without BRAF mutation. N Engl J Med. 2014;372(4):320-330.

VOL. 11, NO. 2 THE AMERICAN JOURNAL OF HEMATOLOGY/ONCOLOGY 33
· commentary ·
The Evolving Role of Surgery in Advanced Melanoma
Richard Essner, MD, FACS
T he recent rapid development of targeted drugs and im-munotherapies for melanoma has left many unanswered questions about the role of surgery in advanced mela-
noma. Traditionally, patients with advanced melanoma were treated first with aggressive surgery. Now, however, the pace of recent developments means that clinical trial data on the role—if any—and timing of surgery in advanced melanoma are lacking.
Take, for instance, BRAF-mutated melanoma, which occurs in approximately 50% of melanomas. In the past, we’d operate on these patients; now we treat them with BRAF inhibitors and/or one of the new immunotherapies, and the role of surgery for these patients has not yet been defined. In fact, except in cases in which a patient is bleeding, needs pain alleviation, or has a tumor that is eroding through the skin, surgery for metastatic disease is rare.
Does this mean that surgery has no place in the future treat-ment of advanced melanoma? The answer is not clear. Among the questions yet to be answered: How much time should we allow for treatment to “work” with the various new therapies? Should a neoadjuvant approach be taken, in which patients are treated with targeted therapy before or during surgery? Will tar-geted or immunotherapy work better after surgery to clear some of the disease? If a patient has a complete response while on ther-apy, do we need to operate to clear residual disease? The clinical trials have not yet caught up to these issues, so right now the an-swers are still the choice of the medical oncologist and surgeon.
Currently, many patients prefer not to have surgery, because treatment with one of the new therapies means potentially less morbidity, less time lost from work, and no surgical wound. However, the majority of patients aren’t cured by the new treat-ments. Some patients may remain on drug therapy for years, trying successive drugs for their melanoma. The problem with this approach is that in the process of trying different therapies, the melanoma may grow or spread. By the time a patient has run out of drug options, he or she may have numerous tumors and no longer be a candidate for surgery. It’s not clear if surgery at some point in the process of trying drug therapy would help prevent the spread or growth of the disease, but there is much room for uncertainty. As an example, trials of anti-PD-1 antibody
therapy have demonstrated a lower success rate in patients with more tumor volume. This begs the question of whether surgery to remove some of the tumors would improve the success rate of the anti-PD-1 antibody in these patients.
New therapies are also changing the role of surgery for in-tran-sit melanoma. Clinical trials are exploring intralesional injection of the experimental agents talimogene laherparepvec (T-VEC) and PV-10 (from Provectus). Use of BRAF inhibition, anti-CT-LA-4 therapy, or immune checkpoint blockade with a PD-L1 inhibitor are also under consideration. These strategies may impact the role of surgery in management of in-transit disease. However, as with management strategies for other forms of ad-vanced melanoma, clinical trials are needed to better define if and how surgery can be used to optimize outcomes.
Affiliations: Richard Essner, MD, FACS, is adjunct professor, Division of Oncology, UCLA, Jonsson Comprehensive Can-cer Center and co-director, Melanoma Program, Cedars-Sinai Health Center, in Los Angeles, CA. Disclosure: Dr Essner reports no relevant financial conflicts of interest to disclose. Address correspondence to: Richard Essner, MD, FACS, Ce-dars-Sinai Health Center, Los Angeles, CA 90025.

34 www.ajho.com February 2015
· perspective ·
Is There an Optimal Intersection for Targeted and Immunotherapy Treatments for Melanoma?
Jason J. Luke, MD, FaCP
IntroductionThe clinical management of patients with BRAF-mutant ad-vanced melanoma has undergone a dramatic transition over the past several years with the development of targeted kinase inhib-itor approaches as well as immune-checkpoint blockade. Since 2011, 6 drugs have obtained FDa approval for the treatment of patients with advanced melanoma. These drugs include ipilim-umab, the fully human monoclonal antibody against cytotoxic T-lymphocyte antigen 4 (CTLa-4); vemurafenib and dabrafenib, both serine-threonine kinase inhibitors targeting the mutant protein braFV600; the MeK1/2 kinase inhibitor trametinib; and
the anti-programmed death 1 (PD-1) antibody pembrolizumab. The first 4 of these drugs have been shown to improve the over-all survival (OS) of patients with melanoma in phase 3 clinical trials,1-5 and data for pembrolizumab are maturing.6
The PD-1 antibody nivolumab, approved in December 2014, also has shown an improvement in OS compared with dacarba-zine chemotherapy in patients with BRAF wild-type melanoma,7 and the combination of nivolumab with ipilimumab appears to improve response rates more than anti-PD-1 antibodies alone.8 additionally, the MeK1/2 inhibitor cobimetinib has improved OS when administered in combination with vemurafenib.9 In considering these treatment options for a patient, it is clear that the response rate of kinase inhibitors targeting mutant BRAF is high; however, response duration is limited in most patients. Ipilimumab has a lower response rate and longer time to treat-ment benefit, but has been associated with approximately 20% survival at 3 years and potentially long-term survival thereafter in patients without disease progression at that point.10 anti-PD-1 antibodies can achieve substantial response rates (21%-40%)6,7,11 with relatively rapid onset and durability of responses, while an-ti-PD-1 with anti-CTLa-4 may be even more robust.
both targeted and immunotherapeutic treatments are import-ant in the management of BRAF-mutant melanoma; however, the optimal sequence and combination of the available treatment agents is not clear. because of the substantial antitumor activity in BRAF-mutant melanoma, inhibition of the mitogen-activated protein kinase (MaPK) pathway, through braF and MeK, may be particularly attractive to combine with immunotherapy. It has been hypothesized that antigen release through tumor cell death mediated by MaPK pathway inhibition may lead to increased antigen presentation or cross-presentation to tumor-specific T cells.12 Inhibition of braF has also been found to increase the expression of melanoma differentiation antigens and induce in-filtration of CD8+ T cells in posttreatment melanoma tumor samples.13 additionally, inhibition of braF and MeK in mela-noma cells leads to increased tumor-specific T cell function, as well as dendritic cell function, in vitro.14 Possible drawbacks of these kinase inhibitors when combined with CTLa-4 or PD-1/
Abstract
Melanoma therapeutics have undergone massive changes
with the approval of BRAF and MEK kinase inhibitors and
immune-checkpoint blocking antibodies against CTLA-4
and PD-1. Targeted and immunotherapy have different
strengths and weaknesses, but both are essential to
clinical management of patients with advanced mela-
noma. Kinase inhibitors have rapid and high response
rates, though their benefit is modest in terms of progres-
sion-free survival. Immunotherapy generally has low-
er response rates and can manifest atypical treatment
kinetics. However, immunotherapy may offer a more
robust potential for durable tumor control. Overall sur-
vival has been improved by both approaches, and next
steps in clinical research will focus on how to combine
these modalities. Early combination clinical trials have
suggested that a cautious approach is appropriate when
combining kinase inhibitors with immunotherapy. To fur-
ther explore this, rigorous biomarker-driven clinical trials
are essential. Beyond just melanoma, it seems likely that
both combinations and sequenced approaches of target-
ed and immunotherapies will be required to harness the
full potential of these approaches for treatment of cancer.
Keywords: BRAF, CTLA-4, MEK, melanoma, PD-1

VOL. 11, NO. 2 THe aMerICaN JOurNaL OF HeMaTOLOGy/ONCOLOGy 35
Is There an OpTImal InTersecTIOn fOr TargeTed and ImmunOTherapy TreaTmenTs fOr melanOma?
programmed death ligand-1 (PD-L1) blockade may include the emergence of resistance and the potential for dampening of the immune response.15
understanding the effects of kinase inhibitors on the immune response is thus essential for the development of rational com-bined targeted-immunotherapeutic drug regimens. Independent of immunotherapy, the combination of braF-MeK inhibition has predominately displaced single-agent braF inhibitor treat-ment, and it is therefore critical to delineate the impact of both a braF inhibitor and MeK inhibitor on the immune response.
Differential Immune Effects of Kinase InhibitorsThere is increasing evidence that kinase inhibitors exert effects on the immune system in addition to the tumor cells they are designed to target. The specific mechanisms by which this occurs are variable. Some examples include impact on the T cell recep-tor through Lck inhibition,16 blockade of cytokine production through modulation of Src,17 and suppression of myeloid-derived suppressor cells through KIT.18 Multitarget kinase inhibitors (eg, imatinib) have been shown to have effects on broad immune cell populations such as CD4+ and CD8+ T cells,19 natural killer (NK) cells,20 and dendritic cells.21 These effects can be positive or negative on the development of an antitumor response, with a productive example being observed in gastrointestinal stromal tumors, where imatinib has been shown to activate CD8+ T cells and induce regulatory T cell apoptosis by reducing tumor-cell expression of indoleamine 2,3-dioxygenase.22 This effect was aug-mented by CTLa-4 blockade.
Inhibition of braFV600 by vemurafenib and dabrafenib ap-pears to have relatively little effect on other kinases, and in vitro/in vivo modeling has suggested that braF inhibition improves T lymphocyte recognition of melanoma antigens and increases the numbers of CD4+ and CD8+ tumor infiltrating lymphocytes ob-served in melanoma tumors.23 Other effects of braF inhibition include reduced expression of immunosuppressive cytokines,24 increased activity of adoptively transferred T cells in mouse xeno-grafts,25 and decreased serum tumor necrosis factor-α.26
In contrast to braFV600e inhibition, MeK inhibition may have a negative impact on T-cell function. Notably, MeK inhibition at high doses has been shown to decrease proliferation and viability of T lymphocytes13 (Figure). MeK inhibition also significantly decreases T-cell associated interferon-γ, whereas braF inhibi-tors do not. Further, MeK inhibition enhances the expression of forkhead box P3 (FoxP3)-positive T cells,27 favoring T cell anergy and potentially contributing to an immunosuppressive tumor microenvironment. Finally, MeK inhibition, but not braF in-hibition, negatively impacts dendritic cell cytokine secretion and decreases antigen presentation.14
a caveat to the impact on T cells by MeK inhibitors may be when they are used in combination with a braF inhibitor. In
a small series of patient samples, no significant differences in quality of T cell infiltrate could be observed between patients receiving single-agent braF inhibitor versus braF-MeK inhibi-tor combination.28 This may be due to the paradoxical activation of the MaPK pathway observed after administration of a braF inhibitor.29 Whereas braF inhibitors block erK signaling in BRAF-mutant melanoma, braF inhibitors have the opposite effect in BRAF wild-type immune cells, where they cause hyper-activation of erK signaling. This can lead to enhancement of T-cell activation and may overcome the inhibitory effect of MeK inhibitors.30 Thus, further research is needed to define the role of MeK inhibitors in combination with immunotherapy.
Clinical ExperiencePreclinical data investigating the intersection between targeted and immunotherapies is developing and may guide future ap-proaches. In the meantime, some lessons can be learned from studies already completed. Though initial reports are limited, the incidence of toxicity has been higher than expected. In renal cell carcinoma (rCC), the vascular endothelial growth factor re-ceptor (VeGFr) inhibitor sunitinib was combined with tremeli-mumab, a CTLa-4 monoclonal antibody.31 In this study, 9 of 28 patients with rCC experienced clinically significant toxicity,
FIGURE. Effects of BRAF and MEK Inhibitors on Immune Cells
MEK inhibitionDendritic cell
Dendritic cell
T cell
T cell
- No negative effect on T cell or DC function- Increased tumor infiltration with CD4 and CD8 cells in vivo
Possibly negative effecton T cell or DC function
MDA
MDA BRAF inhibition
Tumor antigens from necroticmelanoma available for cross presentation to DCs
Tumor Cell
BRAF-mutant melanoma releases suppressive factors including cytokines and other molecules. Treatment with MEK or BRAF inhibitors increases the expression of melanoma differentiation antigens (MDA) and increases their presentation by antigen presenting cells such as dendritic cells (DCs). Whereas BRAF inhibition does not appear to have negative effects on immune cells, MEK inhibition may limit immune function and activation.

36 www.ajho.com February 2015
· perspective ·
predominantly transaminitis and acute renal failure. Prelimi-nary data have been presented regarding the combination of the multitarget (including VeGFr) kinase inhibitors sunitinib and pazopanib with nivolumab, revealing improvement in response rates relative to historical controls but significant toxicity, with treatment being discontinued due to toxicity in 36% and 25% of patients, respectively.32 These toxicities were also predominately hepatic and renal in nature.
In melanoma, the combination of kinase inhibitor and im-mune-checkpoint blockade has also been noted to manifest higher-than-expected toxicity. In the phase 1 study of vemu-rafenib and ipilimumab, 6 of 10 patients experienced dose-lim-iting immune-mediated hepatitis, leading to the combination being abandoned in clinical practice.33 a similar phase 1 study of vemurafenib in combination with the anti-PD-L1 antibody MPDL3280a had to be modified to a staggered dosing approach for the 2 agents due to dose-limiting transaminitis.34 The com-bination of dabrafenib and trametinib with ipilimumab was also discontinued due to the appearance of more severe colitis with bowel perforation. It did appear that dabrafenib without trametinib could be given safely with ipilimumab, though mon-itoring for immune-related hepatitis was recommended.35 based on these and other studies, hepatitis appears to be a common toxicity when combining targeted and immunotherapies.
The mechanisms of augmented immune-related adverse events seen with combination therapies are controversial. It is unknown whether these are related to kinase inhibitors or im-mune-checkpoint blockade. although some of these combina-tions have shown encouraging clinical benefit, care will need to be given in developing combinations of kinase inhibitors with inhibitors of immune checkpoints, especially as anti-CTLa-4 with anti-PD-1 has also shown increased toxicity.8
Discussionremarkable advances with kinase inhibitors and immune check-point blockade have ushered in a new era in melanoma ther-apeutics. Combined braF and MeK inhibition yields rapid responses in most patients; however, the duration of benefit remains modest. Immune checkpoint blockade by ipilimumab, and possibly anti-PD1 antibodies, may offer a greater potential for durable disease control, though the response rates are lower and atypical treatment responses can be difficult to manage in clinic. In this context, there is hope that synergies between these fundamentally different approaches can be identified and trans-lated into improved antitumor activity. based on the experience of combination approaches to date, cautious and prudent evalu-ation will be necessary given the documented increased toxicity in early studies. This will be especially important as further com-binations come into investigation such as PD-1 blockade with other immunomodulators (cytokines or other immune check-point inhibitors), VeGF inhibition, and chemotherapy.
In clinical practice, the appropriate choice of frontline therapy is guided by limited evidence. Many experts in the field suggest that consideration of immunotherapy initially is most prudent because, if the immune response is induced, it may be that no further therapy is needed. This is in contrast with targeted ther-apy, where treatment is open-ended and will almost always even-tually fail. Some have also proposed that patients treated with targeted therapy initially have poorer outcomes with immuno-therapy as second-line treatment, though data supporting this are preliminary.36
Moving forward, there are several studies evaluating con-comitant or sequential administration of kinase inhibitors and immune checkpoint blockade. examples include a study of braF-MeK inhibition with an anti-PD-1 antibody (dabrafenib, trametinib, pembrolizumab) in BRAF-mutant melanoma (Clini-calTrials.gov Identifier: NCT02130466), a study of the MeK in-hibitor cobimetinib with the anti-PD-L1 antibody MPDL3280a in NRAS-mutant melanoma (ClinicalTrials.gov Identifier: NCT01988896), as well as sequential approaches of initial braF plus MeK (dabrafenib-trametinib) combination as compared with initial anti-CTLa-4 plus anti-PD-1 (ipilimumab-nivolumab) combination with eventual crossover to the alternate approach (ClinicalTrials.gov Identifier: NCT02224781).
Drug development in molecular and immunotherapy has set a new bar in advanced melanoma, and this paradigm is likely to become relevant to all cancers in the near future. The best approach in combining these therapies is an open question that urgently needs to be answered. as research moves forward, in-corporation of the underlying tumor genotype will likely need to be taken into account given the emerging data that mutation in NRAS may increase response to immunotherapy.37 Optimal sequencing and dosing of targeted and immunotherapy will only be elucidated in clinical trials, and the development of thera-peutic biomarkers will be critical. It seems likely that both com-bination and sequential approaches of kinase inhibitors with immunotherapy will be required depending on the molecular biology of the patient’s tumor (BRAF vs NRAS vs other) and the particular immunotherapy under study.
Affiliation: Jason J. Luke, MD, FaCP, is assistant professor of Medicine at the university of Chicago.Disclosure: Dr Luke has served as a consultant or on paid advi-sory boards for Genentech, amgen, and bayer.Acknowledgment of Funding Support: Paul Calabresi Career Development in Clinical Oncology award (5K12Ca139160-05)Address correspondence to: Jason J. Luke, MD, FaCP, 5841 S. Maryland ave. MC2115, Chicago, IL 60637. Phone: (773) 834-3096; fax: (773) 702-0963; email: [email protected]

VOL. 11, NO. 2 THe aMerICaN JOurNaL OF HeMaTOLOGy/ONCOLOGy 37
Is There an OpTImal InTersecTIOn fOr TargeTed and ImmunOTherapy TreaTmenTs fOr melanOma?
REFEREnCES1. Chapman Pb, Hauschild a, robert C, et al. Improved survival with vemurafenib in melanoma with braF V600e mutation. N Engl J Med. 2011;364:2507-2516.2. Hauschild a, Grob JJ, Demidov LV, et al. Dabrafenib in braF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358-365.3. Flaherty KT, robert C, Hersey P, et al. Improved survival with MeK inhibition in braF-mutated melanoma. N Engl J Med. 2012;367:107-114.4. Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711-723.5. robert C, Karaszewska b, Schachter J, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib [published online November 16, 2014]. N Engl J Med. www.nejm.org/doi/full/10.1056/NeJMoa1412690. accessed January 22, 2015.6. Hamid O, robert C, Daud a, et al. Safety and tumor respons-es with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369:134-144.7. robert C, Long GV, brady b, et al. Nivolumab in previous-ly untreated melanoma without braF mutation [published online November 16, 2014]. N Engl J Med. www.nejm.org/doi/full/10.1056/NeJMoa1412082. accessed January 22, 2015.8. Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122-133.9. Larkin J, ascierto Pa, Dreno b, et al. Combined vemurafenib and cobimetinib in braF-mutated melanoma. N Engl J Med. 2014;371:1867-1876.10. Schadendorf D, Hodi F, robert C, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipili-mumab in metastatic or locally advanced, unresectable melano-ma. Presented at: the european Cancer Congress 2013; Septem-ber 27-October 1, 2013; amsterdam, The Netherlands. abstract Lba24. 11. Topalian SL, Sznol M, McDermott DF, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014;32(10):1020-1030.12. Hong DS, Vence L, Falchook G, et al. braF(V600) inhibitor GSK2118436 targeted inhibition of mutant braF in cancer pa-tients does not impair overall immune competency. Clin Cancer Res. 2021;18:2326-2235.13. boni a, Cogdill aP, Dang P, et al. Selective braFV600e inhibition enhances T-cell recognition of melanoma without af-fecting lymphocyte function. Cancer Res. 2010;70:5213-5219.14. Ott Pa, Henry T, baranda SJ, et al. Inhibition of both braF
and MeK in braF(V600e) mutant melanoma restores com-promised dendritic cell (DC) function while having differential direct effects on DC properties. Cancer Immunol Immunother. 2013;62:811-822.15. Jiang X, Zhou J, Giobbie-Hurder a, et al. The paradoxical activation of MaPK in melanoma cells resistant to braF inhi-bition promotes PD-L1 expression that is reversible by MeK and PI3K inhibition. Clin Cancer Res. 2012;19(3):598-609.16. Schade ae, Schieven GL, Townsend r, et al. Dasatinib, a small-molecule protein tyrosine kinase inhibitor, inhibits T-cell activation and proliferation. Blood. 2008;111:1366-1377.17. Weichsel r, Dix C, Wooldridge L, et al. Profound inhibi-tion of antigen-specific T-cell effector functions by dasatinib. Clin Cancer Res. 2008;14:2484-2491.18. Kao J, Ko eC, eisenstein S, et al. Targeting immune suppress-ing myeloid-derived suppressor cells in oncology. Crit Rev Oncol Hematol. In press.19. Ozao-Choy J, Ma G, Kao J, et al. The novel role of tyrosine kinase inhibitor in the reversal of immune suppression and mod-ulation of tumor microenvironment for immune-based cancer therapies. Cancer Res. 2009;69:2514-2522.20. Kreutzman a, Juvonen V, Kairisto V, et al. Mono/oligoclo-nal T- and NK-cells are common in chronic myeloid leukemia patients at diagnosis and expand during dasatinib therapy. Blood. 2010;116(5):772-782.21. ray P, Krishnamoorthy N, Oriss Tb, et al. Signaling of c-kit in dendritic cells influences adaptive immunity. Ann New York Acad Sci. 2010;1183:104-122.22. balachandran VP, Cavnar MJ, Zeng S, et al. Imatinib potenti-ates antitumor T cell responses in gastrointestinal stromal tumor through the inhibition of Ido. Nat Med. 2011;17:1094-1100.23. Wilmott JS, Long GV, Howle Jr, et al. Selective braF in-hibitors induce marked T cell infiltration into human metastatic melanoma. Clin Cancer Res. 2011;18(5):1386-1394.24. Khalili JS, Liu S, rodriguez-Cruz TG, et al. Oncogenic braF(V600e) promotes stromal cell-mediated immunosuppres-sion via induction of interleukin-1 in melanoma. Clin Cancer Res. 2012;18:5329-5340.25. Liu C, Peng W, Xu C, et al. braF inhibition increases tu-mor infiltration by T cells and enhances the antitumor activity of adoptive immunotherapy in mice. Clin Cancer Res. 2013;19:393-403.26. Hong DS, Vence L, Falchook G, et al. braF(V600) inhibitor GSK2118436 targeted inhibition of mutant braF in cancer pa-tients does not impair overall immune competency. Clin Cancer Res. 2012;18(8):2326-2335.27. Gabrysova L, Christensen Jr, Wu X, et al. Integrated T-cell receptor and costimulatory signals determine TGF-beta-depen-dent differentiation and maintenance of Foxp3+ regulatory T cells. Eur J Immunol. 2011;41:1242-1248. 28. Tompers Frederick D, Piris a, Cogdill aP, et al. braF inhi-

38 www.ajho.com February 2015
· perspective ·
bition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res. 2013;19(5):1225-1231.29. Poulikakos PI, Zhang C, bollag G, et al. raF inhibitors transactivate raF dimers and erK signalling in cells with wild-type braF. Nature. 2012;464:427-430.30. Callahan MK, Masters G, Pratilas Ca, et al. Paradoxical ac-tivation of T cells via augmented erK signaling mediated by a raF inhibitor. Cancer Immunol Res. 2014;2:70-79.31. rini bI, Stein M, Shannon P, et al. Phase 1 dose-escalation trial of tremelimumab plus sunitinib in patients with metastatic renal cell carcinoma. Cancer. 2011;117:758-767.32. amin a, Plimack e, Infante Je, MS, et al. Nivolumab (an-ti-PD-1; bMS-936558, ONO-4538) in combination with suni-tinib or pazopanib in patients (pts) with metastatic renal cell carcinoma (mrCC). J Clin Oncol. 2014;32:5s (suppl; abstr 5010). 33. ribas a, Hodi FS, Callahan M, et al. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med. 2013;368:1365-1366.34. Hamid O, Sosman J, Lawrence D, et al. Clinical activity, safe-ty, and biomarkers of MPDL3280a, an engineered PD-L1 anti-body in patients with locally advanced or metastatic melanoma (mM). J Clin Oncol. 2013;31(suppl; abstr 9010). 35. Puzanov I, Callahan M, Linette G, et al. Phase 1 study of the braF inhibitor dabrafenib (D) with or without the MeK inhibitor trametinib (T) in combination with ipilimumab (Ipi) for V600e/K mutation–positive unresectable or metastatic mel-anoma (MM). J Clin Oncol. 2014;32:5s(suppl; abstr 2511).36. ackerman a, Klein O, McDermott DF, et al. Outcomes of patients with metastatic melanoma treated with immunotherapy prior to or after braF inhibitors. Cancer. 2014;120(11):1695-1701.37. Joseph rW, Sullivan rJ, Harrell r, et al. Correlation of NraS mutations with clinical response to high-dose IL-2 in pa-tients with advanced melanoma. J Immunother. 2012;35:66-72.

VOL. 11, NO. 2 THE AMERICAN JOURNAL OF HEMATOLOGY/ONCOLOGY 39
· perspective ·
Radiation and Melanoma: A Phoenix Rising
Stephen L. Shiao, MD, PhD, and Omid Hamid, MD
Radiation therapy (RT) plays an integral role in the treatment of many cancers; however, for patients with melanoma, many oncologists do not consider radiation as a treatment option. This is due mainly to the outdated perception that melanomas are “resistant” to radiation. Much of this misperception is based on data using historical techniques and radiation doses that have long since been abandoned. Fortunately, the advent of new mo-dalities of highly-focused RT, the increasing understanding of the role of the immune system in regulating the response to RT, and the recent development of a multitude of immune-oncologic treatment modalities should, and will, change the role of RT in the treatment of all stages of melanoma.
Highly-focused RT refers to a group of techniques including intensity modulated radiation therapy (IMRT), 3D conformal radiation therapy (3DCRT), and stereotactic radiation therapy (SRT), which allow for dramatic dose escalation due to the ability of these techniques to shape the radiation dose around normal dose-limiting tissue structures. Early trials suggesting that high-dose-per-fraction RT (hypofractionated RT) may be effective in melanoma focused on the treatment of brain metastases. Ste-reotactic radiosurgery (SRS) techniques allowed for large single doses of RT (eg, 24 Gy in a single fraction) to be delivered with limited toxicity, and retrospective data examining outcomes in
melanoma brain metastases have shown local control rates of approximately 70% to 80%.1,2 Other trials in melanomas of the head and neck in the postoperative setting have shown similar results with short courses of high doses per fraction RT (5-7 Gy).3
The main issue with hypofractionated RT is that for normal tissues it is associated with an increase in late radiation toxicity, particularly fibrosis. Modern advances in the delivery of RT have allowed for significantly more precise targeting throughout the body such that the amount of normal tissue for a given treatment can often be dramatically reduced. Current techniques such as stereotactic body radiotherapy (SBRT), similar to SRS for the brain, have allowed for the effective treatment of metastatic mel-anoma, with local control rates of >75% in locations such as spine, lung, and liver that previously would have been left un-treated or treated to suboptimal doses.4
In addition to technical advances in the delivery of RT, there has been a recent breakthrough in the understanding of the role of the immune system in the biology of the response to RT. Specifically, the immune system has been found to play a critical role in mediating the efficacy of RT in multiple tumor types, including melanoma. Initial evidence from mouse mod-els has demonstrated that the efficacy of RT depends in part on radiation triggering an antitumor immune response. These models revealed that RT generates an immune response by re-leasing pro-inflammatory molecules, such as type I interferons,5 high-mobility group protein B1 (HMGB1),6 and calreticulin,7 that are sensed by the immune system, leading to the develop-ment of an antitumor response consisting largely of antitumor cytotoxic CD8+ T cells.8,9 Absence of any of the immune compo-nents involved in this process leads to significant reduction in the efficacy of RT on a tumor. It remains unknown whether similar immunologic mechanisms operate in humans, and more studies collecting post-treatment tumor and peripheral blood specimens are needed to understand the immunologic events occurring in patients. Nevertheless, the promising experimental data has led many scientists and clinicians to explore strategies to target vari-ous pathways in the immune system in combination with RT in an attempt to improve the efficacy of both treatments.
With the advent of checkpoint inhibitors and the emerging
Abstract
Although radiation therapy is a mainstay in the treatment
of many cancers, many oncologists do not consider ra-
diation as a treatment option for melanoma due to the
outdated perception that melanomas are “resistant” to
radiation. Fortunately, the advent of new modalities of
radiation therapy allowing for precise targeting of high
doses of radiation therapy and the recognition that these
higher doses can synergize with emerging immunomod-
ulatory agents such as the checkpoint inhibitors have
changed, and will continue to expand, the role of radia-
tion therapy in the treatment of melanoma.
Key words: radiation, immunotherapy, melanoma

40 www.ajho.com FEBRUARY 2015
· perspective ·
concept that the effect of high-dose per fraction RT is not only due to ablation of the irradiated site but also due to the poten-tial to promote immune responses, combining RT with immu-notherapy has become an important new avenue for investiga-tion. Experimental evidence from animal models suggests that in melanoma and other cancers, RT can potentiate the effect of anticancer immunotherapies, such as dendritic cell vaccines10 and checkpoint blockade.11 In these settings, hypofractionated RT seems to be the critical component, as human trials with standard RT and immunomodulatory agents such as high-dose IL-2 did not have similar efficacy.12 Intriguingly, these studies of hypofractionated RT and immunomodulatory agents suggest that in addition to improving local control, RT may also lead to a systemic anticancer immune response coined the abscopal effect.13
Although rare, the idea that RT in combination with immu-nomodulatory agents can be a systemic treatment has generated intense interest from researchers in radiation biology and immu-nology.11 Early trials in melanoma using RT and immunothera-peutics have shown tremendous promise in terms of both local control and systemic responses, suggesting that RT in combina-tion with cytokines or checkpoint inhibitors can be synergistic in terms of generating an antitumor immune response.14,15 Seung and colleagues, in a phase 1 trial of SRT and high-dose IL-2, found a systemic response rate of 71.4%14 compared with the 15% to 20% response rates reported for high-dose IL-2 alone.16,17 Responses in these patients were associated with an increase in CD4+ effector memory T cells.14 A similar dramatic systemic response was observed with ipilimumab and SRT in the case report by Postow et al who also found corresponding increases in activated CD4+ T cells and decreases in the myeloid-derived suppressor cells (MDSC), an inhibitory immune cell thought to suppress antitumor immune responses.13 Mechanistically, animal models and human correlates have revealed that RT can alter the immunogenicity of tumors, making them more susceptible to im-mune-based targeting.14,18 Current and future trials are testing the parameters for treating patients with RT and immunotherapeu-tic combinations in melanoma and a multitude of other tumor types.19,20 Critical questions that need to be addressed in these trials include the timing of RT and immunotherapy and whether this timing differs by the agent, whether the synergy between RT and checkpoint inhibitors also applies to other immunothera-peutics that target other pathways, and, finally, what are the best conditions for RT delivery (site of irradiation, fraction size, and number) to optimize the immunologic response. Participation in the ongoing and future trials studying RT and immunotherapy will help address these questions and, in so doing, may change the standard of care for our patients with melanoma and other advanced malignancies.
Thus, despite the historical concept of melanoma as a radiore-sistant tumor, new paradigms that employ RT to treat melanoma are rapidly emerging. New techniques employing higher doses
of RT have proven to be effective tools in the treatment of brain metastases and other metastatic sites in patients with melano-ma. These new techniques that are allowing for higher ablative doses of RT to be employed, along with increasing evidence that RT can act as a powerful immunomodulatory agent, will make RT an essential consideration in future treatment algorithms for melanoma.
Affiliations: Stephen L. Shiao, MD, PhD, is from the Depart-ment of Radiation Oncology, Cedars-Sinai Medical Center, Los Angeles, CA. Omid Hamid, MD, is from the Angeles Clinic and Research Institute, Los Angeles, CA.Disclosures: Dr Shiao has no conflicts of interest to declare. Dr Hamid is a speaker and consultant for, and receives research funding from, Genentech, Inc. Address correspondence to: Stephen L. Shiao, MD, PhD, De-partment of Radiation Oncology, Cedars-Sinai Medical Center, 8700 Beverly Blvd, Los Angeles, CA 90047. Phone: (310) 423-8077; fax: (310) 423-6161; email: [email protected]
RefeRences1. Radbill AE, Fiveash JF, Falkenberg ET, et al. Initial treatment of melanoma brain metastases using gamma knife radiosurgery: an evaluation of efficacy and toxicity. Cancer. 2004;101(4):825-833.2. Selek U, Chang EL, Hassenbusch SJ, 3rd, et al. Stereotactic radiosurgical treatment in 103 patients for 153 cerebral melanoma metastases. Int J Radiat Oncol Biol Phys. 2004;59(4):1097-1106.3. Ang KK, Byers RM, Peters LJ, et al. Regional radiotherapy as adjuvant treatment for head and neck malignant melanoma. Preliminary results. Arch Otolaryngol Head Neck Surg. 1990;116(2):169-172.4. Jahanshahi P, Nasr N, Unger K, et al. Malignant melanoma and radiotherapy: past myths, excellent local control in 146 studied lesions at Georgetown University, and improving future management. Front Oncol. 2012;2:167.5. Deng L, Liang H, Xu M, et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity. 2014;41(5):843-852.6. Apetoh L, Ghiringhelli F, Tesniere A, et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13(9):1050-1059.7. Obeid M, Tesniere A, Ghiringhelli F, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13(1):54-61.8. Lugade AA, Sorensen EW, Gerber SA, et al. Radiation-induced IFN-gamma production within the tumor microenvironment influences antitumor immunity. J Immunol. 2008;180(5):3132-

VOL. 11, NO. 2 THE AMERICAN JOURNAL OF HEMATOLOGY/ONCOLOGY 41
Radiation and MelanoMa: a Phoenix Rising
3139.9. Lugade AA, Moran JP, Gerber SA, et al. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J Immunol. 2005;174(12):7516-7523.10. Lim JYH, Brockstedt DG, Lord EM, Gerber SA. Radiation therapy combined with Listeria monocytogenes-based cancer vac-cine synergize to enhance tumor control in the B16 melanoma model. Oncoimmunology. 2014;3(6):e29028.11. Barker CA, Postow MA. Combinations of radiation thera-py and immunotherapy for melanoma: a review of clinical out-comes. Int J Radiat Oncol Biol Phys. 2014;88(5):986-997.12. Lange JR, Raubitschek AA, Pockaj BA, et al. A pilot study of the combination of interleukin-2-based immunotherapy and radiation therapy. J Immunother. 1992;12(4):265-271.13. Postow MA, Callahan MK, Barker CA, et al. Immunologic correlates of the abscopal effect in a patient with melanoma. N Engl J Med. 2012;366(10):925-931.14. Seung SK, Curti BD, Crittenden M, et al. Phase 1 study of stereotactic body radiotherapy and interleukin-2--tumor and im-munological responses. Sci Transl Med. 2012;4(137):137ra74.15. Barker CA, Postow MA, Khan SA, et al. Concurrent radio-therapy and ipilimumab immunotherapy for patients with mela-noma. Cancer Immunol Res. 2013;1(2):92-98.16. Rosenberg SA, Lotze MT, Muul LM, et al. Observations on the systemic administration of autologous lymphokine-activated killer cells and recombinant interleukin-2 to patients with meta-static cancer. N Engl J Med. 1985;313(23):1485-1492.17. Atkins MB, Lotze MT, Dutcher JP, et al. High-dose recombi-nant interleukin 2 therapy for patients with metastatic melano-ma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17:2105-2116.18. Sharabi AB, Nirschl CJ, Kochel CM, et al. Stereotactic radi-ation therapy augments antigen-specific PD-1 mediated anti-tu-mor immune responses via cross-presentation of tumor antigen [published online December 19, 2014]. Cancer Immunol Res. pii: canimm.0196.2014.19. Crittenden M, Kohrt H, Levy R, et al. Current clinical trials testing combinations of immunotherapy and radiation. Semin Radiat Oncol. 2015;25(1):54-64.20. Demaria S, Pilones KA, Vanpouille-Box C, et al. The optimal partnership of radiation and immunotherapy: from preclinical studies to clinical translation. Radiat Res. 2014;182(2):170-181.

42 www.ajho.com february 2015
CME
Dates of Certification: February 20, 2015- February 20, 2016
Medium: Print with online posttest, evaluation, and request for credit
Medical WriterKathleen KraftonDisclosure: No relevant financial relationships with commercial inter-ests to disclose.
The American Journal of Hematology/Oncology Editorial BoardDebu Tripathy, MDProfessor of Medicine and ChairDepartment of Breast Medical OncologyThe University of Texas MD Anderson Cancer CenterHouston, TX Disclosure: Grant/Research Support: Genentech/ Roche, Pfizer, Puma, Inc. (clinical trial support contracted to University of Southern Califor-nia); Consultant: Eisai, Novartis
Myron Czuczman, MD Professor of OncologyChief, Lymphoma/Myeloma Service Department of Medicine Head, Lymphoma Translational Research Laboratory Department of Immunology Roswell Park Cancer Institute Buffalo, NY Disclosure: Other Support: Advisory Board: Algeta, Celgene Corpora-tion, Teva, Boehringer-Ingelheim, Mundipharma.
Staff/Planner Disclosures and Conflict of Interest ResolutionThe staff of PER® (Ann C. Lichti, CCMEP, Megan O’Connell, Allison Muller, PharmD, Michael Perlmutter, PharmD, and Beth Cameron, PhD) as well as the editorial staff of The American Journal of Hematology/Oncology (Devera Pine) have no relevant financial relationships with commercial interests to disclose.
In accordance with ACCME’s Standards for Commercial SupportSM, PER® resolved all conflicts of interest (COI) prior to the release of this CME activity using a multistep process.
Overview
This CME activity features data from 4 presentations at the 2014 San Antonio Breast Cancer Symposium, chosen for their impact on current clinical practice or because they lay the groundwork for further investi-gations. Topics include the treatment of premenopausal breast cancer, early evaluation of immunotherapy in the treatment of triple negative breast cancer, outcomes from the ICE trial concerning the treatment of elderly patients with early-stage breast cancer, and the first presenta-tion of a checkpoint inhibitor in breast cancer.
Target Audience
This activity is directed toward medical oncologists who treat patients with breast cancer. Fellows, nurses, physician assistants, nurse practi-tioners, and other healthcare providers may also participate.
Learning Objectives
After participating in this CME activity, learners should be better pre-pared to:1. Review recent data on the treatment of breast cancer presented at
national society meetings2. Evaluate emerging breast cancer clinical data regarding new agents
and evolving strategies
Accreditation/Credit Designation
Physicians’ Education Resource®, LLC, is accredited by the Accreditation Council for Continuing Medical Education to provide continuing medical education for physicians.
Physicians’ Education Resource®, LLC, designates this enduring ma-terial for a maximum of 1.0 AMA PRA Category 1 CreditTM. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
No commercial support was received for this CME-certified activity. This activity was funded entirely by PER®. Instructions for Participation/How to Receive AMA PRA Category 1 CreditTM
1. Read the article in its entirety.2. Use the QR Code or type http://www.gotoper.com/LINK/140 into your Web browser to access the posttest.3. Complete and pass the posttest with a score of 70% or higher.4. Complete the evaluation and request for credit. Participants may immediately download a CME certificate upon successful completion of these steps.
Off-Label Disclosure and Disclaimer
This CME activity may or may not discuss investigational, unapproved, or off-label use of drugs. Participants are advised to consult prescribing information for any products discussed. The information provided in this CME activity is for continuing medical education purposes only, and is not meant to substitute for the independent medical judgment of a physician relative to diagnostic and treatment options for a specific patient’s medical condition.
DisclaimerThe opinions expressed in the content are solely those of the individual faculty members and do not reflect those of PER®.
Contact information for questions about the activity:Physicians’ Education Resource®, LLC666 Plainsboro Road, Suite 356Plainsboro, NJ 08536Phone: (888) 949-0045E-mail: [email protected]
recap of SabCS 2014: Changes for Today and Hope for Tomorrow

VOL. 11, NO. 2 THe aMerICaN JOurNaL Of HeMaTOLOGy/ONCOLOGy 43
breast cancer
The 2014 San antonio breast Cancer Symposium (SabCS) took place December 9-13 at the Henry b. Gonzalez Convention Center in San antonio, Texas, with more than 7000 in attendance. each year, this event draws leading researchers and clinicians from around the world for a presentation of the latest information geared to basic, translational, and clinical cancer research professionals.
Key developments reported at SabCS 2014 included practice-changing data for the treatment of premenopausal breast cancer, early evaluation of immunotherapy in the treatment of triple-negative breast cancer, outcomes from the ICe trial concerning the treatment of elderly patients with early-stage breast cancer, and the first presentation of a checkpoint inhibitor in breast cancer.
In this CMe activity, we review a select group of abstracts from SabCS 2014, chosen for their impact on current clinical practice or because they lay the groundwork for further investigations, followed by expert commentary from Debu Tripathy, MD, chair, breast medical oncology at MD anderson Cancer Center in Houston, Texas.
Updates in the Treatment of Premenopausal Breast CancerOne of the most noteworthy and practice-changing presentations at SabCS 2014 was results of the large international Suppression of Ovarian function Trial (SOfT). To date, tamoxifen has been the hormonal therapy of choice following surgery for premeno-pausal women with breast cancer. However, data from SOfT in-dicate that ovarian function suppression (OfS) combined with endocrine therapy may convey a significant advantage for a sub-set of women in this patient population.1
beginning in November 2003 and extending to January 2011, 3047 premenopausal women with estrogen receptor (er)+ and/or progesterone receptor (Pr)+ breast cancer were randomized to 1 of 3 arms: 5 years of tamoxifen with or without OfS (via the GnrH agonist triptorelin, oophorectomy, or irradiation) or the aromatase inhibitor exemestane plus OfS. The trial was stratified by the use of prior chemotherapy: 47% of participants had received no prior chemotherapy while 53% received prior chemotherapy. all patients were premenopausal, which was con-firmed by estradiol levels within 8 months of completion.1
The trial’s primary objective was the comparison of tamoxi-fen alone versus tamoxifen plus OfS when tested at a 2-sided 0.05 level with median follow-up of at least 5 years; a secondary objective was to compare tamoxifen with exemestane plus OfS. The study’s primary endpoint was disease-free survival (DfS), and secondary endpoints included breast cancer-free interval (bCfI), distant recurrence-free interval (DrfI), and overall sur-vival (OS).1
after a median follow-up of 67 months, the 5-year rate of DfS was 86.6% in the tamoxifen plus OfS group versus 84.7% in the tamoxifen arm (hazard ratio [Hr], 0.83; 95% CI, 0.66-1.04; P = .10). The primary analysis in the overall population was not statistically significant (Hr, 0.78; 95% CI, 0.62-0.98; P = .03). OS data are not mature, but the 5-year rate of OS was 96.7% for tamoxifen plus OfS and 95.1% for tamoxifen (Hr, 0.74; 95% CI, 0.51-1.09; P = .13).1
among patients with no prior chemotherapy, bCfI was >95% with tamoxifen alone. There was a 4.5% absolute improvement
in 5-year bCfI with tamoxifen plus OfS versus tamoxifen mono-therapy in patients who remained premenopausal after chemo-therapy. five-year OS in the chemotherapy cohort was 94.5% for tamoxifen plus OfS and 90.9% for tamoxifen alone (Hr, 0.64; 95% CI, 0.42-0.96).1
Grade 3 or higher toxicities were reported for 31% of pa-tients in the tamoxifen plus OfS arm compared with 24% in the tamoxifen-only group. Menopausal symptoms, depression, osteoporosis, hypertension, and diabetes occurred more fre-quently with tamoxifen plus OfS.1
The researchers concluded that the addition of OfS to tamox-ifen did not provide benefit in the overall premenopausal popu-lation after a median 67 months of follow-up. However, among the women who received adjuvant chemotherapy and remained premenopausal, as well in in women under 35 years (the majority of whom received chemotherapy), the addition of OfS reduced disease recurrence and enabled the use of aromatase inhibitor treatment, which further reduced recurrence in these higher risk cohorts.1
Targeting Triple Negative Breast CancerTriple-negative breast cancer (TNbC) is the umbrella term used to define breast cancers with the absence of er, Pr, and human epidermal growth factor receptor 2 (Her2) expression. TNbC has the poorest prognosis of all breast cancer subtypes.2 One of the challenges inherent in treating TNbC is its heterogeneity: 6 distinct TNbC subtypes, each with its own biological composi-tion, were recently identified in gene expression analyses, raising the question of how best to treat, or target, each particular sub-type.3
Data from the Triple Negative breast Cancer Trial (TNT) con-cerning the choice of chemotherapeutic agent in TNbC further informs therapeutic selection in BRCA-mutation positive meta-static TNbC. TNT was designed as a superiority trial to evaluate the use of the platinum agent carboplatin versus docetaxel in patients with metastatic or recurrent locally advanced TNbC or BRCA1/2-mutated breast cancer (N = 376). eligible patients were

44 www.ajho.com february 2015
CME
those with TNbC, or those known to have a BRCA mutation.4 Patients were randomized to carboplatin (target area under
the concentration versus time curve at 6 mg/mL per minute) or docetaxel (100 mg/m2) every 3 weeks for 6 cycles or until dis-ease progression. The study’s primary endpoint was objective response rate (Orr) defined as the proportion of patients with complete or partial response at cycle 3 or 6. Secondary endpoints included progression-free survival (PfS), Orr (crossover treat-ment), OS, and toxicity. a total of 188 patients were randomized to carboplatin (median age: 55.7 years), and 188 were random-ized to docetaxel (median age: 54.9 years).4
In the randomized treatment population, the 2 treatment arms performed similarly, with an Orr of 31.4% for carboplatin and 35.6% for docetaxel (absolute difference –4.2%; 95% CI, –13.7 to 5.3; P = .44). Similarly, no difference in median PfS and OS was demonstrated. Median PfS was 3.1 months in the carbopla-tin arm (95% CI, 2.5-4.2) and 4.5 months in the docetaxel arm (95% CI, 4.1-5.2). Median OS was 12.4 months with carboplatin (95% CI, 10.4-15.3) compared with 12.3 months with docetaxel (95% CI, 10.5-13.6).4
a prespecified subgroup analysis revealed important differenc-es among patients with BRCA1/2 mutations. In this cohort (n = 43), Orr was 68.0% for carboplatin versus 33.3% for docetaxel (absolute difference, 34.7%; 95% CI, 6.3-63.1; P = .03).4
although the trial provided no evidence of superior response in unselected TNbC, the investigators concluded results from TNT support BRCA1/2 genotyping to inform therapeutic selec-tion because carboplatin-treated patients with BRCA1 or BRCA2 mutations demonstrated improved response and PfS compared with docetaxel in the trial.
Immunotherapy for TNBCThe potential role of immunotherapy in the treatment of TNbC is an area of substantial clinical interest. The discovery of a TNbC subtype characterized by elevated expression of immune genes suggests that immune-based therapies may be of benefit to some patients with TNbC.2 Preliminary findings presented at SabC 2014 from a phase 1b study of the anti-PD-1 agent pembrolizumab in TNbC—the first presentation of a checkpoint inhibitor in breast cancer—support further development of this compound in this setting of advanced TNbC.
The PD-1 receptor-ligand pathway can be used by tumors to evade immune surveillance, thereby allowing neoplastic growth.5 Pembrolizumab is a humanized IgG4/kappa, high affinity anti-PD-1 antibody; when pembrolizumab is blocking the inter-action of the inhibitory PD-1 receptor on T cells with its ligands PD-L1 and PD-L2 expressed on some tumor cells, the antibody prevents this method of tumor evasion from being effective.
Nanda and colleagues reported on their multicenter, nonran-domized trial of pembrolizumab administered intravenously at 10 mg/kg2 every 2 weeks in patients with recurrent or metastat-ic PD-L1-positive TNbC.5 The primary study objectives were to determine the safety, tolerability, and antitumor activity of pem-
brolizumab in this setting. Secondary objectives included assess-ments of PfS, OS, and duration of response.5
a total of 32 patients were enrolled in this ongoing study; the median age of patients was 51.9 years. The majority of patients had disease that had progressed on several lines of treatment used in the setting of advanced disease.5
Preliminary analysis of data collected in November 2014 indi-cates that 1 patient had a complete response, 14.8% of patients had a partial response, 25.9% of patients had stable disease, and 44.4% of patients had progressive disease.5
Overall, 56.3% (18/32) experienced an adverse event (ae) of any grade. Grade 3 aes included anemia, headache, aseptic men-ingitis, and pyrexia (n = 1 each). One patient experienced a grade 4 ae of decreased blood fibrinogen. One fatality occurred due to disseminated intravascular coagulation with thrombocytopenia; this was the only treatment-related ae leading to drug discontin-uation.5 further studies in TNbC are planned for pembrolizum-ab as well as other checkpoint inhibitors.
Treating Early Breast Cancer in Postmenopausal Womenbest practices for the treatment of elderly patients with early-stage breast cancer has been the subject of some debate. This has been complicated by the underrepresentation of elderly women in clinical trials, despite the fact that approximately 50% of newly diagnosed breast cancers occur in women older than 65 years.6
The prospective, multicenter, randomized phase 3 ICe study was designed to investigate if capecitabine added to the bisphos-phonate ibandronate would lead to improved outcomes com-pared with ibandronate alone in elderly breast cancer patients with moderate- and high-risk primary breast cancer who were not considered candidates for standard chemotherapy.6
The controlled, open-label trial enrolled female patients 65 years or older with unilateral or bilateral breast cancer classified as either node-positive or high-risk node-negative (tumor size ≥2 cm, grade >I, and/or ER- and PR-negative) and who had a Charlson Comorbidity Index (CCI) of 2 or less. Patients received either ibandronate alone for 2 years (50 mg orally daily or 6 mg intravenously every 4 weeks) or the same ibandronate regimen together with capecitabine (2000 mg/m²) on days 1 to 14 every 3 weeks for 6 cycles. Treatment was initiated within 6 months following axillary dissection. Patients with hormone-sensitive dis-ease received an endocrine therapy according to guidelines. The primary objective was invasive DfS.6
The trial was held in Germany between June 2004 and au-gust 2008; 1358 patients were randomized and treated (681 in the ibandronate arm and 677 in the ibandronate/capecitabine arm) and the median patient age was 71 years for both treatment groups.6
results were similar among treatment arms: at 3 years, DfS was 85.4% in the capecitabine plus ibandronate arm versus 84.3% in the ibandronate arm.6 at 5 years, 78.8% of patients in the capecitabine plus ibandronate arm were free of invasive disease, compared with 75% of patients in the ibandronate alone

VOL. 11, NO. 2 THe aMerICaN JOurNaL Of HeMaTOLOGy/ONCOLOGy 45
breast cancer
arm. Grade 3/4 toxicities in the capecitabine group were 31% compared with 8.7% in the ibandronate group.6 The investiga-tors concluded that the ICe study failed to substantiate that ad-juvant capecitabine improves invasive DfS in ibandronate-treat-ed patients.
REfERENCES1. francis Pa, regan MM, Gleming Gf, et al. randomized com-parison of adjuvant tamoxifen (T) plus ovarian function suppres-sion (OfS) versus tamoxifen in premenopausal women with hor-mone receptor-positive (Hr+) early breast cancer (bS): analysis of the SOfT trial. Presented at: San antonio breast Cancer Sympo-sium; December 9-13, 2014; San antonio, TX. abstract S3-08. 2. Stagg J, allard b. Immunotherapeutic approaches in tri-ple-negative breast cancer: latest research. Ther Adv Med Oncol. 2013;5(3):169-181.3. Mayer Ia, abramson VG, Lehmann bD, et al: New strategies
for triple-negative breast cancer—deciphering the heterogene-ity. Clin Cancer Res. 2014;20:782-790.4. Tutt a, ellis P, Kilburn L, et al. The TNT Trial: a randomized phase III trial of carboplatin (C) compared with docetaxel (D) for patients with metastatic or recurrent or locally advanced triple negative or brCa 1/2 breast cancer (CruK/07/012). Present-ed at: San antonio breast Cancer Symposium; December 9-13, 2014; San antonio, TX. abstract S3-08.5. Nanda r, Chow LQ, Dees eC, et al. a phase Ib study of pem-brolizumab (MK-3475) in patients with advanced triple-negative breast cancer. Presented at: San antonio breast Cancer Sympo-sium; December 9-13, 2014; San antonio, TX. abstract S1-09. 6. Von Minckwitz G, reimer T, Potenberg J, et al. The phase III ICe study: adjuvant ibandronate with or without capecitabine in elderly patients with moderate or high risk early breast cancer. Presented at: San antonio breast Cancer Symposium. December 9-13, 2014; San antonio, TX. abstract S3-04.
AJHO: Data from the oral abstract, A phase Ib study of pembroli-zumab (MK-3475) in patients with advanced triple-negative breast can-cer, were expected to have the potential to be practice changing. Did the data live up to expectations? What are the major impli-cations of this study? Dr Tripathy: The phase 1b study of pembrolizumab is important because it’s the first study to test a new type of immunotherapy known as checkpoint blockade. It is blockade of the PD-1 or PD-L1 proteins, which are involved in suppressing the immune system and preventing autoimmunity. However, when you block these so-called checkpoint proteins, you can also enhance im-munity against cancer. To date, most of the work in checkpoint blockade has been in melanoma and renal cell cancer; in fact, pembrolizumab is approved in melanoma. This was the first study that looked at checkpoint blockade in breast cancer.
There has been a lot of interest in TNbC because there are some signs that some cases of TNbC are immunogenic, as evi-denced by the presence of lymphocyte infiltrates. TNbC is also a difficult disease to treat, with chemotherapy being the only op-tion, so this was a logical setting for a phase 1b study.
I believe that the study did live up to its expectations. It wasn’t as dramatic as we had hoped for, but about one-fifth of patients had a response. Keep in mind, we have never had a targeted ther-apy that has led to any responses in TNbC as a single agent, and many of the patients in this group had already undergone several
lines of chemotherapy for advanced disease. There are some general next steps for this approach to therapy.
first, we need to better define the optimal group of patients for this type of therapy. In this study, the researchers selected pa-tients who had more than 1% expression of PD-L1 in either the stromal cells or the tumor. The question is, should we be looking at other proteins to help us better identify patients who already have some baseline amount of immunity and thus might benefit more from this drug?
The other area that I think is even more important is treating patients at the right stage of disease. I think the time that immu-notherapy works best is when the overall burden of disease in the body is lower, not in patients with bulky advanced disease, but perhaps in the adjuvant setting and in patients who have a very high risk of recurrence. These types of trials are already under consideration and being designed.
I think we are just beginning in this area and I look forward to more information about this exciting line of therapy. Whether it’s going to ultimately live up to the expectations and actually prevent recurrences or have significant impacts on survival, of course, remains to be seen.
AJHO: another important trial reported on at SabCS was the TNT trial (The TNT trial: A randomized phase III trial of carbopla-tin (C) compared with docetaxel (D) for patients with metastatic or
Expert CommentaryDebu Tripathy, MDChair, Breast Medical Oncology DepartmentMD Anderson Cancer CenterHouston, Texas

46 www.ajho.com february 2015
CME
recurrent locally advanced triple negative or BRCA1/2 breast cancer [CRUK/07/012]). What particular implications do you think the findings from TNT might have both in the clinical setting and for future explorations?Dr Tripathy: The TNT trial was another one focused on TNbC. There’s been a lot of excitement about the use of platinum agents in TNbC because they seem to generate a better response than standard agents in the laboratory setting. TNbC can also have what is known as the brCa phenotype: not only germ-line mutations in BRCA, but there may be some cancers with normal BRCA that still have defects on the brCa pathway.
We’ve never formally tested whether or not a platinum agent is better than standard therapy, such as docetaxel, so that is exactly what this study did. The investigators compared carboplatin with docetaxel in patients with advanced TNbC. The study showed that both drugs were about equal in terms of response rates, but when investigators examined the subset of patients that actually did have BRCA mutations, they found that platinum appeared to be superior. That is what many would have expected, but it was very important to formally demonstrate that.
The other thing this trial did is ask the question, might there be patients who have normal BRCA genetics but who still have some abnormalities in the brCa pathway who might also ben-efit from platinum agents? The investigators performed a ho-mologous recombination deficiency (HrD) assay, and what they found is that there was no difference based on the HrD assay. The HrD assay is being developed for clinical use and it may identify a brCa-like group of patients. This study did not sup-port the notion that the HrD assay made a difference; however, we have to recognize that all the tumors tested were from the early-stage cancer and not from recurrence or metastasis.
I think that field is going to need some more work, but what we can say for now is that platinum agents do seem to have pref-erential activity in BRCA-positive cases. The next step is going to be to design trials specifically in that group of patients to see if we should be using those agents in the standard setting. The other important question that is also being asked in ongoing random-ized trials is whether ParP inhibitors should be used for these patients. There’s a large ongoing trial called the Olympia study that is currently enrolling patients with TNbC who are also known to have BRCA mutations; eventually, all patients known to have BRCA mutations will be enrolled. The study will test the addition of the ParP inhibitor, olaparib, for 1 year following the completion of all standard therapy compared with placebo. This is going to be an important study not only for TNbC, but for all brCa-related cancers.
AJHO: Let’s discuss neoadjuvant therapy as a research platform for TNbC. Specifically, what makes it an important avenue for study?Dr Tripathy: This is a very important area that will help us ac-celerate drug development. We know that the response to neo-adjuvant therapy, especially for TNbC, is an important predictor of long-term survival. We’ve known for many years now that pa-tients who have a complete pathologic response have a much bet-
ter outcome—perhaps a recurrence risk of about 10%, whereas in patients who do not achieve a complete pathologic response to neoadjuvant therapy, the risk of recurrence may be as high as 40% or 50%, and even higher in some studies. by looking at the response rates and testing different drugs, we may get a much quicker way to test which drugs are likely to be successful in ei-ther advanced or early-stage breast cancer, so many investigators are now taking advantage of this design and taking patients who do not achieve a complete response to neoadjuvant therapy if they have TNbC and comparing the addition of one treatment or another.
Pembrolizumab is going to be tested in a study of that nature and so are ParP inhibitors, for example, in brCa cancers. you can get an answer on complete pathologic response within 4 to 6 months of therapy as opposed to your typical metastatic trial, where it takes 2 to 3 years for the data to mature. So this gives us a better way to select what drugs should move forward into definitive testing, at a much more rapid pace.
AJHO: results from the TeXT and SOfT trials, which were re-ported at the american Society of Clinical Oncology meeting, were considered practice changing by many, opening up the op-tion of treating premenopausal women with hormone-sensitive breast cancer with a combination of ovarian function suppres-sion and an aromatase inhibitor. How might the SOfT analysis that was presented at SabCS concerning only tamoxifen with or without ovarian function suppression further impact practice for treating oncologists? Dr Tripathy: This is an important study because it’s really the first large-scale study to look at ovarian ablation in addition to standard hormonal therapy. for premenopausal patients with er+ cancers, for early-stage treatment the current standard is tamoxifen for 5 years. More recently, we’ve learned that for high-er-risk patients, 10 years is perhaps better than 5 years.
There have been some studies in the past that looked at the addition of oophorectomy, but they have been relatively small and underpowered and haven’t been that informative. They may have suggested that people under the age of 35 or 40 years might benefit. They’ve also shown that patients who do not achieve cessation of their menstrual periods have a worse outcome. In general, however, those studies never proved whether or not sup-pressing the ovaries adds any benefit.
both the TeXT and SOfT trials were designed to ask 2 ques-tions: 1) Is blockade of ovarian function using gonadotropin analogs helpful for premenopausal patients, and 2) If you do block ovarian function, is it better to use tamoxifen or an aro-matase inhibitor?
earlier, it had been shown that patients do benefit a little bit more from aromatase inhibitors than tamoxifen if the ovaries are suppressed, but the SOfT data presented at the San anto-nio meeting showed that the effect of oophorectomy was not statistically significantly better than not blocking the ovaries. However, there are some important subset analyses that were presented. One is that for high-risk patients who are receiving chemotherapy in addition to hormonal therapy, there did seem

VOL. 11, NO. 2 THe aMerICaN JOurNaL Of HeMaTOLOGy/ONCOLOGy 47
breast cancer
to be a clear benefit from ovarian blockade, particularly when an aromatase inhibitor was used. The other thing the data showed was that in women under the age of 35 years, there appeared to be a clear impact of ovarian ablation. Now, those patients had a higher chance of getting chemotherapy, so it’s unclear if it’s simply the fact that they were younger or that they were receiving chemotherapy and were in a higher risk category that conferred the additional benefit.
I would say the major conclusions of the study are that, at a high level, ovarian ablation does not seem to help the aver-age patients, and certainly not the low-risk patients. In fact, the study showed that low-risk patients, stage I and even stage II, have an excellent overall outcome with tamoxifen alone: 95% or greater survival. for those patients, standard tamoxifen probably suffices. However, for your higher-risk patients, particularly those getting chemotherapy and, even more specifically, those who do not have cessation of their menstrual periods, one may consider ovarian ablation.
I do think that this is going to impact practice for those sub-sets, even though the results of the main trial were negative with respect to oophorectomy in the overall population. I would also say that, for high-risk patients, if you are going to use ovarian ablation, you might as well use an aromatase inhibitor.
AJHO: The potential for improved outcomes versus the risk of toxicity with immunotherapy drug combinations has been the subject of much debate. How do you navigate this divide in clin-ical practice? are there practical strategies clinicians can employ to manage patients who may be at higher risk for immune-medi-ated adverse reactions?Dr Tripathy: for now, this remains a research question in the area of breast cancer because none of these drugs are yet ap-proved. but when we design clinical trials, we have to be very mindful of toxicities. fortunately, the newer generation of im-munomodulatory drugs, the PD-1 and PD-L1 inhibitors, seem to have fewer side effects than the last generation, such as the CTLa4 blockers like ipilimumab, which have a lot of side ef-fects, including mostly skin and gastrointestinal toxicities. The PD-1 and PD-L1 inhibitors do have the same toxicities, but at a much lower rate. as we get more information on the benefit and efficacy of immunotherapy in breast cancer, as well as the short- and long-term toxicities, we’ll be able to answer these questions more precisely.
AJHO: What were some key take-away messages from San an-tonio—data with immediate impact on clinical practice, new hy-potheses for future research, or anything else that stood out to you as the blockbuster finding or an unexpected result?Dr Tripathy: I would say that the most practice-changing infor-mation came from the TeXT and SOfT trials. I wouldn’t call these dramatic or unexpected findings, because we felt that oo-phorectomy probably does have a role, and what we’ve shown is that it has a very borderline role when you look at the overall population, but I do think it’s going to change practice. That’s the one practice-changing set of studies I would say emerged from
San antonio, specifically for the higher-risk patients, particularly those receiving chemotherapy and particularly those under the age of 35 years.
The other area that I would say was groundbreaking was im-munotherapy. While the results were not dramatic, they did show for the first time that we can get a handle on TNbC with relatively safe immunotherapy. It’s just the beginning. We have to understand more about immunotherapy. We have to design immunotherapy trials in the right patient populations, possibly in the adjuvant setting, where immunotherapy can actually have the potential to save lives. So this is just the beginning, but it’s a very exciting beginning.
One thing we are learning as a community is that breast cancer is a collection of smaller entities that each have distinct biologi-cal characteristics. So the importance of us linking tissue-based research to drug-based research is critical. We have to be treating the right target population. We have to be aware of the science and we have to make sure that we gather tissue and continue to develop a robust platform for analysis, at the genomic level, at the protein level, and at the epigenetic level. There’s really a revolution in science going on, but it’s going to be challenging because instead of now just treating breast cancer as 1 group or even the subtypes like triple-negative, we’re realizing that there are even subtypes within these subtypes. So you’re going to see much more about this in the future, and it’s a challenge because now we’re dealing with smaller numbers of patients and we have statistical challenges that make it more difficult to interpret these studies. We have to be creative in how we design trials going for-ward; you will see this in the trials that are reported in the future. They’re going to be smaller, but they’re going to be much more biologically focused.

iNSTRUCTiONS FOR AUTHORS
The rapid pace of discovery in the field of oncology presents practicing oncologists with the difficult challenge of implementing novel research findings into clinical practice. In order to accelerate the translation of academic advances to community-based practice, The American Journal of Hematology/Oncology aims to provide practical interpretations of the latest advances in medical and hematologic oncology and to help practicing oncologists gain a better understanding of how these advances have changed the treatment landscape for both solid and hematologic malignancies. The editors are pleased to consider manuscripts on a wide range of topics related to the journal’s mission. Articles of interest include:• Original research• Reviews
- State of the Art Updates- On the Horizon- Emerging Guidelines
• Editorials and perspectives- Clinical Controversies - Looking Forward- Brief Reports- Pivotal Trials- New Technologies- Meeting Updates - Case Reports- Survivorship Issues- Team Approaches (Allied Health/Care Extenders)
- Pharmacology Updates- Oncology Practice Issues: Evolving
Detailed descriptions of each category can be found in the Instructions for Authors at www.ajho.com.
T h e
TH
E O
FFICIAL J
OU
RN
AL O
F o f
LYMPHOMAThe Role of PET Imaging in the Management of Patients With Hodgkin Lymphoma Craig Moskowitz, MD
LEUKEMIAA Primer on Molecular Testing in Chronic Myeloid Leukemia (PCR for Poets)Jerald P. Radich, MD
OVARIAN CANCERGenomic (“Precision”) Medicine in the Management of Ovarian CancerMaurie Markman, MD PER: 9TH ANNUAL INTERNATIONAL SYMPOSIUM ON OVARIAN CANCER AND GYNECOLOGIC MALIGNANCIES®
What Would Clinicians Do If BRCA or Lynch Positive?Medical Management, Insurance Discrimination, and Confidentiality
A m e r i c a n
J o u r n a l
H e m a t o l o g y /
O n c o l o g y ®
A Peer-Reviewed Resource
for Oncology Education
CMEMedical Crossfire: Recent Advances in the Treatment of Metastatic Melanoma A CME-certified enduring material sponsored by Physicians’ Education Resource®, LLC
ajho
www.AJHO.com
All manuscripts will undergo peer review, and authors of accepted articles will receive an honorarium and will be required to sign an authorship form disclosing any possible conflicts of interest. To submit an article to The American Journal of Hematology/Oncology or if you wish to speak to an editor, please e-mail Devera Pine at [email protected].
Call for Papers
AJHO_CallForPapers_08'14.indd 5 10/7/14 10:53 AM

iNSTRUCTiONS FOR AUTHORS
VOL. 11, NO. 2 THE AMERICAN JOURNAL OF HEMATOLOGY/ONCOLOGY 49
Mission & Scope The American Journal of Hematology/Oncology is a monthly, peer-reviewed publication that provides original research, reviews, and editorials/commentaries that address cutting-edge developments in genomics, targeted therapies, molecular diagnostics, and pathways related to oncological science and clinical practice. As the official publication of Physicians’ Education Resource (PER), the journal’s mission is to advance cancer care through professional education. The journal aims to provide practical interpretations of the latest advances in medical and hematologic oncology and to help practicing oncologists gain a better understanding of how these advances are changing the treatment landscape for both solid and hematologic malignancies. Articles published in the journal will illustrate successes and failures in clinical practice and provide prac-tical insights into the myriad decisions oncologists face in everyday clinical practice. Readership: The American Journal of Hematology/Oncology circulates to 10,000 practicing oncologists across the country. Our audience includes medical oncologists, hematologists, pathologists, dermatologists, radiation oncologists, and surgical oncologists. Submitting Manuscripts Requirements for all submissions generally conform to the “Uni-form Requirements for Manuscripts Submitted to Biomedical Journals.”1 Our peer review process is blinded, so all identifying information (ie, author names, affiliations, etc) must be removed from the manuscript file before submission. Manuscripts submitted for publication in The American Journal of Hematology/Oncology must not have been published previously (either in whole or in part) nor currently be submitted elsewhere in either identical or similar form. Material posted on the Internet
or disseminated in any other electronic form constitutes prior publication and may not be considered. Previous publication of a small portion of the content of a manuscript does not necessarily preclude its being published in The American Journal of Hematology/Oncology, but the editors need information about previous publica-tion when deciding how to use space in the journal efficiently.
These restrictions on prior publication, however, do not apply to abstracts or poster presentations published in connection with scientific meetings or to working papers that have been posted on the Web to facilitate peer feedback.
Authors must indicate in the cover letter whether any portion of the manuscript has been previously published and are required to submit copies of related publications (either published, in prepara-tion, or submitted), as well as any manuscripts cited as “in press” to the editors for review. Duplicate, redundant, and/or fragmented publications are not permitted. Refer to Chapter 5 of the American Medical Association (AMA) Manual of Style for further information on duplicate publication.2 Authors should also include a statement in the body of the paper that indicates whether the study was approved by an institutional review board. For all papers (when appropriate), a statement confirming that the informed consent of study subjects was obtained should be included in the manuscript.
Articles of Interest The editors are pleased to consider manuscripts on a wide range of topics related to the Journal’s mission. Authors should write for a sophisticated general audience and recognize that many of The American Journal of Hematology/Oncology readers are not researchers. In addition to evaluating articles for scientific merit, the editors will also assess the overall relevance of the work to the journal’s audience.
If you are uncertain of an article’s appropriateness for The American Journal of Hematology/Oncology, we encourage authors to

iNSTRUCTiONS FOR AUTHORSiNSTRUCTiONS FOR AUTHORS
50 www.ajho.com FEbRUARY 2015
send an abstract or outline of an article to the Editorial Office ([email protected]) to facilitate a pre-submission review with the Editor-in-Chief.
Submissions generally fall into one of the following categories: (1) original research; (2) reviews; or (3) editorials or perspectives.
Original research articles should employ a clear hypothesis-driven research question and an appropriate research design and analysis to report clinically relevant outcomes. Articles should be 2000-2500 words (excluding abstract, references, tables, etc) and contain no more than 5 graphic elements. Supplemental data (extra tables, fig-ures, or appendices) will be made available on the journal’s website at the time of publication. Authors should indicate what material is intended as Web-only content and include the appropriate refer-ence or callout in the text to these Web-exclusive elements.Reviews should provide concise, up-to-date reviews of novel therapies and treatment strategies or other clinically relevant over-views. Authors should present real-world examples and discussion of the inherent challenges of incorporating new therapeutics, new treatment strategies, and new diagnostic tools into clinical practice. Articles should be 1500 to 2000 words with at least 1 graphic ele-ment to illustrate a key concept. The journal’s graphic design staff is available to develop original figures based on a sketch provided by authors. Types of review articles are as follows:• State-of-the-Art Update: Reviews of the evidence support-
ing recent key developments in the treatment of cancer, with a particular focus on information essential and applicable to clinical practice. Please illustrate key points with tables and/or figures (assistance is available from the journal staff for the development of figures).
• On the Horizon: Reviews of translational research, therapies, and technology that are in development but that clinicians will need to be aware of within the next few years. If applicable, please illustrate key points with figures (assistance is available from the journal staff) and provide relevant citations.
• Emerging Guidelines: Highlights of the key points of the most recent clinical practice guidelines, with expert perspec-tives/opinions on the changes to the guidelines. This can be 1000 words or less, without a figure.
Editorials and perspectives can employ several formats that provide concise and lively discussions on timely and relevant topics. These would typically involve areas of rapid change, controversy, or new areas that have the potential for major future clinical impact in oncology. These should be brief (less than 1500 words) with appropriate citations. Examples include:• Clinical Controversies: Opinion pieces that discuss relevant
and controversial issues in oncology (eg, maintenance ritux-imab and its role in indolent lymphoma, should DCIS be considered a cancer, when to intervene or start chemotherapy in prostate cancer, what is the quality of life impact of PFS
vs OS improvements, etc). In some cases, two authors would contribute opposing but coordinated (pro/con, or point/counterpoint) pieces.
• Looking Forward: New areas of research or clinical care that are not well known to many oncologists, but may in the future impact cancer care or research directions. This perspective would be a “thought piece” without significant amounts of data or citations.
• Brief Reports: Brief and topical perspectives and updates on new concepts, treatments, and diagnostic assays (less than 1000 words)
• Pivotal Trials: Summaries of clinical trials of interest. Should include the background/rationale, eligibility, treatment sche-ma, contact information, and NCT link (up to 1000 words)
• New Technologies: Discussions of imaging and tissue-based technologies, genomics, bioinformatics, etc (up to 1000 words)
• Meeting Updates: Summaries of presentations at key CME meetings, conferences, and congresses, with expert perspec-tives on the reported findings (please query the editorial team first to avoid duplication of coverage of meetings)
• Case Reports: Unusual cases, situations, exceptional respond-ers, including histology and imaging
• Survivorship: Discussions of survivorship topics and symp-tom management (1000-1500 words)
• Allied Health/Care Extenders: Discussions of how to best use a team approach; this can be a case report format—eg, discussion of how an individual team met and overcame a challenge or streamlined a process to improve patient care using allied health professionals/care extenders (1000-1500 words). The journal’s editors encourage allied health profes-sionals on the oncology care team to author or co-author these articles
• Pharmacology Updates: Brief overview of new drugs—mechanisms, dosing, side effects, drug interactions (1000-1500 words). These could be contributed by a pharmacist or a PharmD and may have the look of a write-up typical of a Pharmacy & Therapeutics Committee formulary application.
• Oncology Practice Issues: Evolving aspects of oncology practice such as insurance coverage, electronic medical re-cords, quality assurance, accelerated drug approvals, survivor-ship, and patient education/communication that present new perspectives and useful information to oncologists (1000-1500 words)
Authorship Only persons who have made a direct contribution to the content of a paper should be listed as authors. The number of authors listed with the manuscript should not exceed 10; more than 10 requires written justification and approval from the Editor-in-Chief.
The American Journal of Hematology/Oncology uses the criteria provided by the “Uniform Requirements for Manuscripts Submit-

iNSTRUCTiONS FOR AUTHORS
VOL. 11, NO. 2 THE AMERICAN JOURNAL OF HEMATOLOGY/ONCOLOGY 51
ted to Biomedical Journals”1 to determine authorship. Each author should have participated sufficiently in the work to take public responsibility for the content. Authorship credit should be based only on substantial contributions to (a) conception and design, or analysis and interpretation of data; and to (b) drafting the article or revising it critically for important intellectual content; and on (c) final approval of the version to be published. Conditions (a), (b), and (c) must all be met.1
Individuals who have contributed to a paper but who do not meet the criteria for authorship should be acknowledged. Disclosures It is our policy to have all authors disclose relationships with any commercial interest that may present a real or perceived conflict of interest if: (a) the relationship is financial and occurred within the past 12 months; and (b) the author discusses products or ser-vices of that commercial interest. Relevant financial relationships are those relationships in which the author (and/or the author’s spouse or partner) benefits in any dollar amount by receiving a salary, royalty, intellectual property rights, consulting fee, honoraria, ownership interest (eg, stocks, stock options, or other ownership interests, excluding diversified mutual funds), or other financial benefit. Financial benefits are usually associated with roles, such as employment, management position, independent contractor (including contracted research), consulting, speaking and teaching, membership on advisory committees or review panels, board mem-bership, and/or other activities for which remuneration is received or expected. In addition, authors are required to report all financial and material support for their research, which includes (but is not limited to) grant support and funding sources and any provision of equipment or supplies. To this end, all authors must read and sign the journal’s Author Disclosure Form.
The name of the organization funding or initiating a research project should be made explicit on the title page (eg, “This study was funded by the XYZ Corporation.”). Relevant financial relation-ships (whether direct to the authors or through a third party) for research and/or writing, including funding, grants, honoraria, etc, must also be named on the title page. If the funding organization had any role in the collection of data, its analysis and interpreta-tion, and/or in the right to approve or disapprove publication of the finished manuscript, this must be noted in the cover letter and described in the text. The editorial staff may inquire further about financial disclosure after the manuscript is submitted. If the manuscript is accepted for publication, disclosure statements will be printed as part of the published paper. ManuscriptSpecifications Manuscript components (cover letter, text, tables, figures, related papers, etc) must be included as part of the submission process. All manuscripts should include the following components:Cover Letter: A cover letter must accompany each submission and
include any background information about the submission (ie, how it contributes to the existing literature, whether any portion has been previously presented or published, etc) that would aid in the editors’ initial evaluation. Include a statement that the manuscript has been read and approved by all authors.Titles. Titles should be concise (fewer than 10 words) and stimu-late reader interest. Provide a brief running title in addition to the main article title.
The title page should include the following information:• the complete manuscript title and subtitle, if any• the full names of each author, followed by their highest aca-
demic degree• the name, address, telephone, fax, and e-mail information of
the corresponding author• the institutional affiliations for each author at the time the
work was completed• a concise summary of the article to appear in the table of
contents (no more than 25 words) • practical application of your work (a bulleted list that high-
lights the real-world impact of your work)• indication of the source of funding (including grant numbers,
grant agencies, corporations, or sponsors)• the number of pages, references, figures, and tables• a word count (excluding references, tables, and figures)
Abstract. An abstract is required for all manuscript submissions. The abstract should not exceed 250 words and should summarize the salient data and the principal conclusion of the piece.
Text. All text should be double-spaced, including the acknowledg-ments, references, tables, and legends. Cite references, tables, and figures in sequential order in the body of the paper. Measurements of length, height, weight, and volume should be reported in metric units. Temperatures should be given in degrees Celsius. Blood pres-sures should be listed in millimeters of mercury. Except for units of measure, abbreviations are discouraged.
Any abbreviation or acronym must be spelled out in full when it first appears in the text, followed by its abbreviation in parentheses. State the generic name (not the trade name) for all drugs.
Permissions: Data and/or figures reproduced from another pub-lished source must be properly cited and acknowledged. Authors are required to obtain written permission from the appropriate author and/or copyright holder to reproduce previously published or copyrighted material. Authors must also obtain permission from at least 1 author when citing unpublished data, “in-press” articles, and/or personal communications. Copies of permission statements should be included with manuscript submissions.
Acknowledgments. Include a list of acknowledgments, if ap-propriate. Refer to the “Authorship” section for an explanation

iNSTRUCTiONS FOR AUTHORSiNSTRUCTiONS FOR AUTHORS
52 www.ajho.com FEbRUARY 2015
of what constitutes authorship and for guidance in distinguishing contributions that warrant an acknowledgment. The corresponding author must affirm that he/she has received permission to list the individuals in the acknowledgment section (see bottom of Author-ship Form).
References. Begin the reference section on a new page and double-space both within and between reference citations. Number references sequentially in the order cited in the text—do not alphabetize. Provide the names of all authors when there are six or fewer; if there are more than six authors, list only the first three authors followed by “et al.” All references must be verified by the authors and should conform to the AMA Manual of Style.2
References cited only in table or figure legends should be numbered in accordance with the sequence established by the first mention of the particular table or figure in the text.
References to papers accepted but not yet published should be designated as “in press” and included in the reference section. Information from manuscripts submitted but not accepted should be cited in the text as “unpublished observations” with written permission from the source. (Include copies of any “in press” and “submitted” manuscripts [ie, papers under consideration at other journals] for the editors’ evaluation as part of your submission.)
Avoid citing “personal communication” unless it provides es-sential information not available from a public source, in which case the name of the person, his or her degree, and the date of communication should be cited in parentheses in the text. Authors should obtain written permission and confirmation of accuracy from the source of a personal communication (see “Permissions” section). Note the format and punctuation in the following sample references:1. Cortes JE, Kim DW, Kantarjian HM, et al. Bosutinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leuke-mia: results from the BELA trial [published online September 4, 2012]. J Clin Oncol. 2012;30(28):3486-3492.2. Wierda WG, O’Brien S. Chronic lymphoblastic leukemia. In: DeVita VT Jr, Lawrence TS, Rosenberg SA, eds. DeVita, Hellman, and Rosenberg’s Cancer: Principles & Practice of Oncology, 9th ed. Phila-delphia: Lippincott Williams & Wilkins; 2011.3. American Cancer Society. Cancer Facts and Figures 2012. http://www.cancer.org/acs/groups/content/@epidemiology-surveilance/documents/document/acspc-031941.pdf Accessed October 5, 2012.
Graphic Elements. Use of graphic elements is strongly encour-aged, and The American Journal of Hematology/Oncology will print up to 5 graphic elements. All supplemental data (eg, appendices and lengthy tables) will be posted on the journal’s website at the time of publication. Authors should indicate what material is intended for web-exclusive content and include the appropriate reference or callout in the text to these web-exclusive elements.
Tables. Place each table on a new page. Number tables sequen-tially in the order they are cited in the text. Include a title for each table. Special characters, abbreviations, and symbols must be explained in a footnote to the table.
Figures. The journal’s production team is available to create figures from sketches provided by the authors. Avoid the use of shading in bar graphs or pie charts—use color or crosshatch pat-terns instead. Number all figures in the order they are mentioned in the text. Any previously published figures must be accompa-nied by written permission from the publisher and/or copyright holder (see “Permissions” section).
Legends. Legends should be double-spaced and include the figure number and a brief description of the illustration. Identify all abbreviations used in the figure at the end of each legend.
Peer Review Each manuscript is sent to the Editor-in-Chief for an internal evaluation to determine its appropriateness. Manuscripts that do not meet the journal’s criteria for overall appropriateness, relevance, originality, and scientific merit will be returned promptly (usually within 2 weeks) so that authors may pursue alternate avenues for publication. Although reviewer selection is ultimately the decision of the edi-tors, authors may provide the names and e-mail information of preferred and nonpreferred peer reviewers. Manuscripts deemed appropriate for The American Journal of Hematology/Oncology will be sent to external peer reviewers. Typically, a manuscript will be sent to a minimum of two reviewers who will be asked to provide feedback on the scientific merit of the paper. The Editorial Office contacts reviewers in advance and asks them to complete their evaluation of a manuscript within two weeks. Re-viewers are asked to treat manuscripts as confidential communica-tions and not to share their content with anyone (except colleagues whom they ask in confidence to assist in reviewing) or to use the content for their own purposes. We do not send a manuscript to a reviewer who is affiliated with the same institution as any of the authors and we ask reviewers to declare any potential conflicts of interest, such as personal ties to an organization with a vested inter-est in the topic of the manuscript. Editorial Decisions We judge manuscripts on the interest and importance of the topic, the intellectual and scientific strength, the clarity of the presenta-tion, and relevance to our readers. We also consider the strength of the paper compared with other papers under review and the number of accepted and previously published papers in the paper’s category. Authors of original research and review articles should take pains to describe exactly how their findings add to the existing literature.

iNSTRUCTiONS FOR AUTHORS
VOL. 11, NO. 2 THE AMERICAN JOURNAL OF HEMATOLOGY/ONCOLOGY 53
The Editorial Office is committed to providing prompt process-ing times and to communicating timely decisions to authors. While the Editorial Office makes every effort to notify authors and keep them informed of any delays, most authors can expect a first deci-sion on their manuscript in approximately 4 to 6 weeks.
We communicate editorial decisions on acceptance or rejection only to the corresponding author. Almost all papers that we accept require some editorial revision before publication.
Accepted Papers Page proofs (PDFs) are e-mailed to the corresponding author before publication. Authors can expect to receive proofs approxi-mately 3 to 4 weeks before the scheduled issue date. All proofs must be returned to the Editorial Office within 48 hours.
References 1. International Committee of Medical Journal Editors. Uniform requirements for manuscripts submitted to biomedical journals: writing and editing for biomedical publication. [Updated April 2010.]http://www.icmje.org/urm_full.pdf. Accessed October 5, 2012.2. Iverson C, ed. Ethical and legal considerations. In: American Medical Association Manual of Style. 10th ed. New York, NY: Oxford University Press; 2007:125-300.
Editorial Offices are located at:The American Journal of Hematology/Oncology Office Center at Princeton Meadows, Bldg 400 Plainsboro, New Jersey 08536E-mail: [email protected]
















![OPTIMAL DECISION SUPPORT MIXTURE MODEL WITH WEIBULL … · 2019-09-28 · Tripathy and Sahoo [18], Tripathy and Sukla [19] developed improved inventory model in fuzzy sense using](https://static.fdocuments.us/doc/165x107/5e5692c04be7281bfb55fd41/optimal-decision-support-mixture-model-with-weibull-2019-09-28-tripathy-and-sahoo.jpg)


